Regelmäßige Trisomie auf Chromosom 18. Diagnose Edwards-Syndrom – was ist das?
Die meisten Frauen wissen genau, was das Down-Syndrom ist. Und während der Schwangerschaft erfahren viele Menschen, dass sehr selten auch eine andere Chromosomenstörung namens Edwards-Syndrom auftritt. Und viele fragen sich, wie man herausfinden kann, wie hoch das Risiko ist, ein Kind mit Edwards-Syndrom zu bekommen, und wie eine solche Pathologie während der Schwangerschaft diagnostiziert wird.
Was ist das Edwards-Syndrom?
Das Edwards-Syndrom ist eine genetische Erkrankung, die durch eine Verdoppelung (Trisomie) des Chromosoms XVIII gekennzeichnet ist und sich durch eine Reihe charakteristischer Missbildungen des Fötus während der Schwangerschaft manifestiert, die häufig zum Tod des Kindes oder zu seiner Behinderung führen. Das heißt, ein Kind entwickelt statt 46 Chromosomen 47 Chromosomen; dieses zusätzliche Chromosom gibt der Krankheit einen anderen Namen – Trisomie 18. Das Syndrom wurde nach dem Forscher John Edwards benannt, der es 1960 erstmals beschrieb.
Warum das Edwards-Syndrom auftritt - Ursachen der Pathologie
Auch wenn die Eltern gesund sind und in der Familie keine solche Pathologie vorliegt, kann jede Frau ein Kind mit Chromosom 18 zur Welt bringen.
Bekanntlich enthält jede menschliche Zelle 46 Chromosomen, weibliche und männliche Keimzellen verfügen über 23 Chromosomen, die bei der Befruchtung der Eizelle zusammengenommen ebenfalls insgesamt 46 Chromosomen ergeben. Beim Edwards-Syndrom sind die Ursachen seines Auftretens unbekannt.
Derzeit ist lediglich bekannt, dass durch bestimmte genetische Mutationen ein zusätzliches 47. Chromosom entsteht (ein zusätzliches Chromosom im 18. Chromosomenpaar, das somit nicht zu 2, sondern zu 3 wird).
In 95 % aller Fälle der Entwicklung des Edwards-Syndroms enthalten die Zellen genau das zusätzliche 18. Chromosom (Trisomie), in 2 % wird jedoch nur eine „Verlängerung“ des 18. Chromosoms (Translokation) beobachtet, während die Gesamtzahl der Chromosomen erhalten bleibt normal und entspricht 46.
In 3 % der Fälle des Edwards-Syndroms kommt es zur „Mosaik-Trisomie“, wenn ein zusätzliches 47. Chromosom im Körper nicht in allen Zellen, sondern nur in einem bestimmten Teil davon gefunden wird. Klinisch verlaufen alle 3 Varianten des Edwards-Syndroms nahezu identisch, die erste Variante kann sich jedoch durch einen schwereren Krankheitsverlauf unterscheiden.
Wie häufig ist diese Pathologie?
Kinder mit Edwards-Syndrom sterben in etwa 60 % der Fälle während der fetalen Entwicklung. Dennoch kommt dieses Syndrom unter den genetischen Erkrankungen bei überlebenden Säuglingen recht häufig vor und tritt in der Häufigkeit nach dem Down-Syndrom an zweiter Stelle auf. Die Prävalenz des Edwards-Syndroms beträgt 1 Fall bei 3.000 bis 8.000 Kindern.
Es wird angenommen, dass diese Krankheit bei Mädchen dreimal häufiger auftritt und das Risiko eines Edwards-Syndroms deutlich steigt, wenn die schwangere Frau 30 Jahre oder älter ist.
Die Sterblichkeitsrate beim Edwards-Syndrom liegt im ersten Lebensjahr bei etwa 90 %, und die durchschnittliche Lebenserwartung bei schweren Fällen der Krankheit beträgt bei Jungen 2–3 Monate und bei Mädchen 10 Monate, und nur wenige überleben das Erwachsenenalter. Am häufigsten sterben Kinder mit Edwards-Syndrom an Erstickung, Lungenentzündung, Herz-Kreislauf-Versagen oder Darmverschluss – Komplikationen, die durch angeborene Fehlbildungen verursacht werden.
Wie äußert sich das Syndrom bei einem Kind?
Die Anzeichen des Edwards-Syndroms können in mehrere große Gruppen eingeteilt werden:
Die erste Gruppe umfasst Symptome, die für das Aussehen des Kindes charakteristisch sind:
- Niedriges Geburtsgewicht (ca. 2.100 – 2.200 Gramm)
- Unverhältnismäßig kleiner Kopf
- Entwicklungsstörungen des Ober- oder Unterkiefers (Mikrognathie)
- Verzerrung der Gesichtsform und Entstehung von Zahnfehlstellungen
- Gaumenspalte (Spalte des harten Gaumens) oder Lippenspalte (Spalte der Oberlippe)
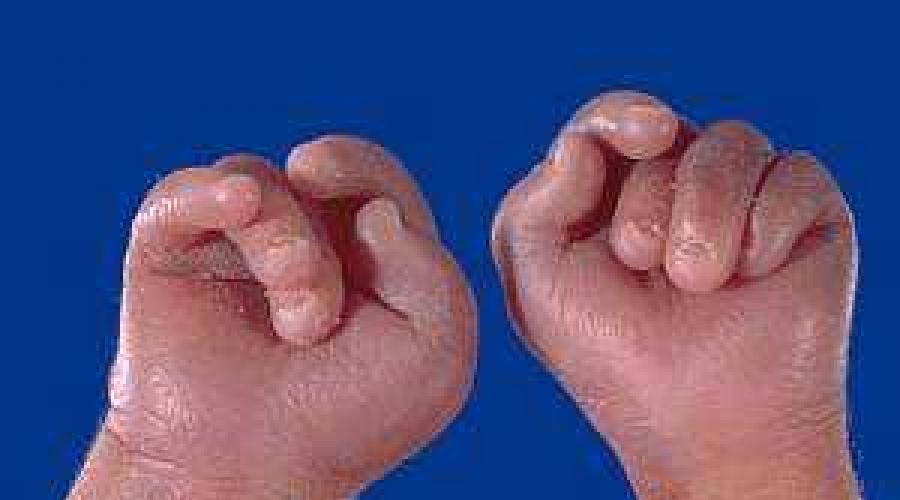
- Die Finger der Hand sind geballt und ungleichmäßig zur Faust positioniert
- Tief angesetzte Ohren
- Schwimmhäute oder vollständige Verschmelzung der Finger der unteren Extremitäten
- Angeborener Klumpfuß
- „Schaukelnder Fuß“
- Relativ kleine Mundspalte (Mikrostomie)
Die zweite Gruppe umfasst Anzeichen einer Fehlfunktion innere Organe, Motorik und neuropsychische Entwicklung:
- Das Vorhandensein angeborener Herzfehler, zum Beispiel offenes Foramen ovale, ventrikulärer Septumdefekt, offener Ductus arteriosus usw.
- Entwicklung von Leisten- oder Nabelbrüchen.
- Verdauungsorgane: gastroösophageale Refluxkrankheit, gestörter Schluck- und Saugreflex, Ösophagusatresie oder Anus, Meckel-Divertikel, Darmerkrankung.
- Zentralnervensystem: verzögerte neuropsychische Entwicklung, geistige Behinderung, Unterentwicklung des Kleinhirns, Corpus callosum, Glättung oder Atrophie der Gehirnwindungen.
- Urogenitalsystem: Kryptorchismus, Hypospadie bei Jungen, Klitorishypertrophie, unterentwickelte Eierstöcke bei Mädchen, unabhängig vom Geschlecht – hufeisenförmige oder segmentierte Niere, Verdoppelung der Harnleiter.
- Strabismus, Skoliose, Muskelschwund.
So erkennen Sie Pathologien während der Schwangerschaft – Diagnose
Das Patau-Syndrom, das Edwards-Syndrom und andere Trisomien lassen sich am besten vor der Geburt des Kindes erkennen. Die pränatale Diagnostik dieses Syndroms erfolgt in der Regel in 2 Stufen:
- Für einen Zeitraum von 11–13 Wochen (Screening, das auf verschiedenen biochemischen Tests bei einer Frau basiert).
- Bestimmung des fetalen Karyotyps bei gefährdeten schwangeren Frauen.
In der 11.–13. Woche wird der Spiegel bestimmter Blutproteine im Blut einer Frau bestimmt: β-hCG (β-Untereinheit des menschlichen Chorionhormons) und Plasmaprotein A, das mit einer Schwangerschaft in Zusammenhang steht. Unter Berücksichtigung dieser Daten und des Alters der Schwangeren wird dann das Risiko, ein Kind mit Edwards-Syndrom zu bekommen, berechnet und eine Risikogruppe schwangerer Frauen gebildet.
Darüber hinaus wird in der Risikogruppe zu einem späteren Zeitpunkt dem Fötus Material entnommen, um eine genaue Diagnose zu stellen: In der 8. bis 12. Woche handelt es sich um eine Chorionzottenbiopsie, in der 14. bis 18. Woche um eine Amniozentese (Untersuchung des Fruchtwassers). nach 20 Wochen - Cordozentese (intrauterine Blutentnahme aus der Nabelschnur des Fötus mit Ultraschallkontrolle). Anschließend wird im resultierenden Material mittels QF-PCR (quantitative Fluoreszenz-Polymerase-Kettenreaktion) das Vorhandensein oder Fehlen eines zusätzlichen 18. Chromosoms bestimmt.
Wenn eine schwangere Frau keinem genetischen Screening unterzogen wurde, dann mehr später Die vorläufige Diagnose des Edwards-Syndroms erfolgt mittels Ultraschall. Weitere indirekte Anzeichen, aufgrund derer zu einem späteren Zeitpunkt ein Edwards-Syndrom vermutet werden kann:
- Das Vorhandensein von Anomalien in der Entwicklung von Knochen und Weichteilen des Kopfes („Gaumenspalte“, Mikrozephalie, tief angesetzte Ohren, „Lippenspalte“ usw.).
- Erkennung von Herz-Kreislauf-Defekten, Urogenitalsystem sowie des Bewegungsapparates.
Diagnostische Anzeichen des Syndroms bei einem Kind
Nach der Geburt eines Kindes sind die wichtigsten diagnostischen Anzeichen für das Vorliegen eines Edwards-Syndroms die folgenden:

Anzeichen eines charakteristischen dermatografischen Musters:
- unentwickelte distale Beugefalte an den Fingern
- das Vorhandensein einer quer verlaufenden Palmarfurche in 1/3 der Fälle
- Bögen an den Fingerspitzen
- Veränderung im Hautmuster der Handfläche: distale Lage des axialen Triradius und Zunahme der Leistenzahl.
Edwards-Syndrom im Ultraschall - Plexus choroideus Zysten
In den frühen Stadien ist das Edwards-Syndrom im Ultraschall äußerst schwer zu vermuten, jedoch zeigen sich bereits in der 12. Schwangerschaftswoche die dafür charakteristischen Symptome indirekter Natur :
- Anzeichen einer Wachstumsbeschränkung des Fötus
- Bradykardie (verminderte fetale Herzfrequenz)
- Omphalozele (Vorliegen einer Bauchhernie)
- Mangelnde Visualisierung der Nasenknochen
- Die Nabelschnur hat eine und nicht zwei Arterien
Außerdem kann Ultraschall Zysten des Plexus choroideus erkennen, bei denen es sich um Hohlräume mit darin enthaltener Flüssigkeit handelt. Sie stellen an sich keine Gefahr für die Gesundheit dar und verschwinden in der 26. Schwangerschaftswoche. Allerdings gehen solche Zysten sehr oft mit verschiedenen genetischen Erkrankungen einher, beispielsweise dem Edwards-Syndrom (in diesem Fall werden Zysten bei 1/3 der Kinder gefunden, die an dieser Pathologie leiden). Wenn solche Zysten entdeckt werden, wird der Arzt die schwangere Frau daher überweisen Frau zur Untersuchung zu einer genetischen Beratung.
Da Kinder mit Edwards-Syndrom selten ein Jahr überleben, zielt die Behandlung zunächst darauf ab, lebensbedrohliche Entwicklungsstörungen zu beheben:
- Wiederherstellung der Nahrungspassage bei Darm- oder Analatresie
- Ernährung durch eine Sonde ohne Schluck- und Saugreflexe
- antibakterielle und entzündungshemmende Therapie bei Lungenentzündung
Bei einem relativ günstigen Krankheitsverlauf erfolgt die Korrektur einiger Anomalien und Fehlbildungen: chirurgische Behandlung der „Gaumenspalte“, Herzfehler, Leisten- oder Nabelbruch sowie symptomatische medikamentöse Behandlung (Verschreibung von Abführmitteln gegen Verstopfung, „Entschäumer“) bei Blähungen etc.) . 
Kinder mit Edwards-Syndrom sind anfällig für Krankheiten wie:
- Mittelohrentzündung
- Nierenkrebs (Wilms-Tumor)
- Lungenentzündung
- Bindehautentzündung
- pulmonale Hypertonie
- Apnoe
- Bluthochdruck
- Stirnhöhlen, Sinusitis
- Infektionen des Urogenitalsystems
Daher umfasst die Behandlung von Patienten mit Edwards-Syndrom eine rechtzeitige Diagnose und Behandlung dieser Krankheiten.
Prognose für das Kind
In den meisten Fällen ist die Prognose ungünstig. Die wenigen Kinder mit Edwards-Syndrom, die das Erwachsenenalter erreichen, haben schwere geistige Behinderungen und benötigen ständige Pflege und Aufsicht. Bei geeigneten Aktivitäten sind sie jedoch in der Lage, auf Komfort zu reagieren, zu lächeln, selbstständig zu essen und auch mit Betreuern zu interagieren, wodurch sie verschiedene Fähigkeiten erwerben.
Edwards-Syndrom

Edwards-Syndrom- eine Chromosomenerkrankung, die durch Trisomie auf Chromosom 18 verursacht wird und mit mehreren Fehlbildungen einhergeht. Das Edwards-Syndrom ist durch besondere phänotypische Anzeichen (dolichozephale Form des Schädels, Mikrophthalmie, Unterentwicklung der Ohren, Mikroretrognathie usw.), Anomalien des Bewegungsapparates, des Herz-Kreislauf-Systems, des Verdauungssystems, des Urogenitalsystems und des Zentralnervensystems gekennzeichnet. Die Diagnose des Edwards-Syndroms kann während der Schwangerschaft (Ultraschall-Screening, invasive Pränataldiagnostik) oder nach der Geburt des Kindes anhand äußerer Anzeichen und zytogenetischer Untersuchungen gestellt werden. Kinder mit Edwards-Syndrom benötigen eine symptomatische Behandlung und gute Pflege.
Edwards-Syndrom

Das Edwards-Syndrom ist eine quantitative Chromosomenaberration, bei der eine teilweise oder vollständige Trisomie des Autosoms 18 auftritt. Das Syndrom wurde nach dem Genetiker J. Edwards benannt, der die Krankheit 1960 ausführlich beschrieb und über 130 symptomatische Defekte identifizierte, die für diese Pathologie charakteristisch sind. Das Edwards-Syndrom ist nach dem Down-Syndrom die zweithäufigste Chromosomenstörung; Die Häufigkeit der Geburten von Kindern mit Edwards-Syndrom liegt bei 1:5000-7000. Ungefähr drei Viertel aller Patienten mit Edwards-Syndrom sind Mädchen; Es wird angenommen, dass die meisten Schwangerschaften mit einem männlichen Fötus mit einem intrauterinen Tod und einem spontanen Abort enden.
Ursachen des Edwards-Syndroms
Die Entstehung des Edwards-Syndroms wird durch Chromosomenanomalien erklärt, die im Stadium der Gametogenese (Ovogenese oder Spermatogenese) oder der Zygotenfragmentierung auftreten und zu einer Erhöhung der Chromosomenzahl des 18. Paares führen. In 80–90 % der Fälle werden zytogenetische Varianten des Edwards-Syndroms durch eine einfache Trisomie 18 dargestellt, seltener durch eine Mosaikform oder unausgeglichene Umlagerungen (Translokationen).
Die Ursache einer vollständigen Trisomie ist die Nichtdisjunktion der meiotischen Chromosomen. In fast allen Fällen ist das zusätzliche Chromosom mütterlichen Ursprungs. Diese Variante des Edwards-Syndroms ist in ihrer Ausprägung die schwerste und hinsichtlich der Prognose ungünstig. Das Auftreten von Mosaiken ist mit der Nichtdisjunktion der Chromosomen im frühen Stadium der Zygotenspaltung verbunden. In diesem Fall enthalten nicht alle fetalen Zellen das zusätzliche Chromosom, sondern nur ein Teil davon. Translokation – die Anheftung eines Teils des 18. Chromosoms an ein anderes Paar kann sowohl während der Reifung der Gameten als auch nach der Befruchtung erfolgen. In diesem Fall enthalten die Körperzellen zwei homologe 18. Chromosomen und ihr zusätzlicher Teil ist an ein anderes Chromosom gebunden.
Wie beim Down-Syndrom ist das Alter der Mutter der wichtigste Risikofaktor für die Geburt eines Kindes mit Edwards-Syndrom. IN in seltenen Fällen Es kann sein, dass die Eltern Träger einer ausgeglichenen Translokation sind.
Symptome des Edwards-Syndroms
Während der Schwangerschaft werden Polyhydramnion, schwache fetale Aktivität, eine kleine Plazenta und eine einzelne Nabelarterie beobachtet. Ein Kind mit Edwards-Syndrom wird mit geringem Körpergewicht (ca. 2170 g) und pränataler Unterernährung während der Vollschwangerschaft oder sogar nach der Schwangerschaft geboren. Bei einigen Kindern wird bei der Geburt Asphyxie diagnostiziert.
Neugeborene mit Edwards-Syndrom weisen charakteristische phänotypische Merkmale auf, die auf diese chromosomale Pathologie hinweisen. Zunächst wird auf die dolichozephale Form des Schädels hingewiesen, bei der die Längsgröße gegenüber der Quergröße überwiegt, niedrige Stirn, hervorstehender Hinterkopf, Mikrognathie, kleiner Mund, Mikrophthalmie. Kinder mit Edwards-Syndrom leiden häufig an Lippen-Kiefer-Gaumenspalten, Epikanthus, Ptosis, Exophthalmus, Strabismus und einem kurzen Hals mit übermäßigen Hautfalten. Typische Ohrdeformitäten sind kleine Ohrläppchen, fehlender Tragus, enge Gehörgänge und tief angesetzte Ohren.
Das Erscheinungsbild von Kindern wird durch für das Edwards-Syndrom charakteristische Skelettdeformationen ergänzt – gekreuzte Finger, verkürztes Brustbein, Rippenanomalien, angeborene Hüftluxation, Klumpfuß, Kippfuß, Syndaktylie der Füße usw. Viele Kinder haben Hämangiome und Hautpapillome.
Beim Edwards-Syndrom liegen mehrere schwere Anomalien in fast allen Körpersystemen vor. Angeborene Herzfehler können durch Defekte der interventrikulären und interatrialen Septen, Aortenisthmusstenose, Transposition der großen Gefäße, Klappendysplasie, Fallot-Tetralogie, abnormale Drainage der Lungenvenen, Dextrakardie usw. dargestellt werden. Beim Edwards-Syndrom ist die Pathologie von Die Entwicklung des Magen-Darm-Trakts kann nachgewiesen werden: Zwerchfell-, Nabel- und Leistenbruch, Meckel-Divertikel, tracheoösophageale Fisteln, Pylorusstenose, Atresie des Ileums und Anus. Die häufigsten Anomalien des Urogenitalsystems bei Kindern mit Edwards-Syndrom sind Hufeisenniere, Hydronephrose, Blasendivertikel, Hypospadie und Kryptorchismus (bei Jungen), Uterus bicornis, intrauterines Septum und Klitorishypertrophie (bei Mädchen).
Fehlbildungen des zentralen nervöses System gekennzeichnet durch das Vorhandensein von Mikrozephalie, Meningomyelozele, Hydrozephalus, Arnold-Chiari-Malformation, Arachnoidalplexuszysten, Kleinhirn- und Corpus-callosum-Hypoplasie. Alle überlebenden Kinder mit Edwards-Syndrom geistige Beeinträchtigung- Oligophrenie im Ausmaß tiefer Dummheit oder Idiotie.
Neugeborene mit Edwards-Syndrom haben Schwierigkeiten beim Saugen, Schlucken und Atmen, weshalb sie auf eine Sondenernährung oder eine langfristige mechanische Beatmung angewiesen sind. Kinder mit Edwards-Syndrom sterben in der Regel im ersten Lebensjahr aufgrund schwerer angeborener Fehlbildungen und damit verbundener Komplikationen (Herz-Kreislauf- und Atemversagen, Lungenentzündung, Darmverschluss usw.).
Diagnose des Edwards-Syndroms
Die wichtigste diagnostische Aufgabe ist die vorgeburtliche Erkennung des Edwards-Syndroms beim Fötus, da diese Pathologie eine medizinische Indikation für einen künstlichen Schwangerschaftsabbruch darstellt. Das Vorliegen eines Edwards-Syndroms kann bei fetalem Ultraschall und Dopplerographie des uteroplazentaren Blutflusses aufgrund indirekter Anzeichen (multiple Anomalien der fetalen Entwicklung, Agenesie der Nabelarterie, kleine Plazenta, Polyhydramnion usw.) vermutet werden.
Die größte diagnostische Bedeutung hat das standardmäßige pränatale Screening, einschließlich einer Blutuntersuchung auf Serummarker: βhCG und PAPP in der 11.–13. Schwangerschaftswoche; βHCG, Alpha-Fetoprotein und freies Östriol in der 20. bis 24. Schwangerschaftswoche.
Bei der Beurteilung des Risikos, ein Kind mit Edwards-Syndrom zu bekommen, werden biochemische und Ultraschall-Screeningdaten, das Gestationsalter, das Alter und das Körpergewicht der Frau berücksichtigt. Schwangeren mit hohem Risiko wird eine invasive Pränataldiagnostik (Chorionzottenbiopsie, Amniozentese, Cordozentese) mit anschließender fetaler Karyotypisierung angeboten.
Bei einer Lebendgeburt eines Kindes mit Edwards-Syndrom ist eine umfassende Untersuchung zur frühzeitigen Erkennung schwerwiegender Fehlbildungen erforderlich. Ein Neugeborenes mit Edwards-Syndrom sollte von einem Neonatologen, Kinderkardiologen, Kinderneurologen, Kinderchirurgen, Kinderorthopäden, Kinderurologen usw. untersucht werden. Die wichtigsten diagnostischen Tests, die bei einem Kind mit Edwards-Syndrom in den ersten Lebensstunden durchgeführt werden sollten sind Echokardiographie, Ultraschall der Bauchhöhlen und Ultraschall der Nieren.
Behandlung des Edwards-Syndroms
Da Entwicklungsanomalien in den meisten Fällen nicht mit dem Leben vereinbar sind, beschränkt sich die Behandlung von Kindern mit Edwards-Syndrom auf die symptomatische Versorgung mit dem Ziel, physiologische Funktionen aufrechtzuerhalten, das Leben zu verlängern und seine Qualität zu verbessern. Die chirurgische Korrektur angeborener Defekte ist in der Regel riskant und ungerechtfertigt.
Da Kinder mit Edwards-Syndrom geschwächt und anfällig für häufige Harnwegsinfektionen, Mittelohrentzündung, Konjunktivitis, Sinusitis, Lungenentzündung usw. sind, benötigen sie eine sorgfältig organisierte Pflege, eine gute Ernährung und eine regelmäßige Überwachung durch einen Kinderarzt.
Prognose und Prävention des Edwards-Syndroms
In allen Fällen ist die Prognose des Edwards-Syndroms äußerst ungünstig: Jungen leben durchschnittlich 2-3 Monate, Mädchen 10 Monate. Nur 10 % der Patienten überleben bis zu 1 Jahr, nicht mehr als 1 % überleben bis zu 10 Jahre. Kinder mit der Mosaikform des Edwards-Syndroms haben relativ gute Überlebenschancen.
Das Risiko, ein Kind mit Edwards-Syndrom zu bekommen, besteht theoretisch bei jedem Ehepaar; Es ist bekannt, dass diese Wahrscheinlichkeit bei älteren Eltern höher ist (für Frauen über 45 Jahre - 0,7 %). Um chromosomale Pathologien beim Fötus rechtzeitig zu erkennen, sollte das vorgeburtliche Screening, das Teil des Schwist, nicht vernachlässigt werden.
Edwards-Syndrom
Unser professionelles Team beantwortet Ihre Fragen
Edwards-Syndrom (Trisomie 18) ist nach dem Down-Syndrom die zweithäufigste Chromosomenstörung. Die Inzidenz des Edwards-Syndroms liegt bei 1:5000–1:7000 Neugeborenen. Mädchen mit Edwards-Syndrom werden dreimal häufiger geboren als Jungen.
Der „Goldstandard“ zur Erkennung von Chromosomenstörungen ist weltweit seit langem und nach wie vor die Methode der Karyotypisierung mit differenzieller Chromosomenfärbung. Mit dieser Methode können Sie den Karyotyp als Ganzes analysieren und große (mindestens 5-10 Millionen Basenpaare) Chromosomenumlagerungen bestimmen. Es weist jedoch eine Reihe von Einschränkungen auf, wie z. B. Arbeitsintensität, Dauer (1-2 Wochen), hohe Anforderungen an die Qualifikation und Erfahrung des die Studie durchführenden Spezialisten sowie in einigen Fällen technische Probleme (unzureichende Menge und Qualität des untersuchten Materials, Mangel an Mitosen oder Kulturwachstum).
Die Methode der quantitativen Fluoreszenz-Polymerase-Kettenreaktion (QF-PCR), die zunehmend zur Diagnose von Aneuploidien, einschließlich des Edwards-Syndroms, eingesetzt wird, weist diese Nachteile nicht auf (Abb. 1). Diese Methode weist eine mit der Standard-Karyotypisierung vergleichbare Zuverlässigkeit auf, ist schneller, kostengünstiger, stellt weniger Anforderungen an die Menge und Qualität des Materials (da sie nicht mit dem Wachstum einer Zellkultur verbunden ist) und ermöglicht die gleichzeitige Analyse einer großen Anzahl von Proben . Allerdings weist die QF-PCR-Methode auch Einschränkungen auf: In Mosaikfällen ist nur der Nachweis eines hochgradigen Mosaiks (ab 20 %) möglich, darüber hinaus kann das Vorliegen seltenerer Chromosomenstörungen, die mit fetalen Fehlbildungen einhergehen können, nicht ausgeschlossen werden. Bei der pränatalen Diagnose des Edwards-Syndroms ist es notwendig, der Mutter zusätzlich zum fetalen Material biologisches Material zur Verfügung zu stellen, um die Möglichkeit eines falsch negativen Ergebnisses aufgrund einer falschen Entnahme von fetalem Material auszuschließen. Die Analyse des fetalen Materials ist innerhalb von drei Arbeitstagen abgeschlossen.
Beim Edwards-Syndrom kommt es zu einer ausgeprägten Verzögerung der pränatalen Entwicklung, Kinder werden mit pränataler Unterernährung geboren (das durchschnittliche Körpergewicht bei der Geburt beträgt 2340 g). Die äußeren Erscheinungsformen des Edwards-Syndroms sind vielfältig (Abb. 2). Die typischsten sind verzögerte psychomotorische Entwicklung, Hypoplasie der Skelettmuskulatur und des Unterhautfettgewebes, angeborene Herzfehler, Anomalien der Gesichts- und Schädelstruktur (Dolichozephalie, Mikrophthalmie, Verkürzung der Lidspalten, niedrige Lage der Ohren, Mikrognathie, Schrägstellung). Kinn), multiple Deformationen der Hände und Füße, Entwicklungsanomalien des Magen-Darm-Trakts, des Urogenitalsystems und des Zentralnervensystems (Spina bifida, Hypoplasie des Corpus callosum und Kleinhirns). Die Lebenserwartung von Kindern ist stark verkürzt: 90 % von ihnen sterben vor ihrem ersten Lebensjahr an den Folgen angeborener Fehlbildungen (Asphyxie, Lungenentzündung, Darmverschluss, Herz-Kreislauf-Versagen).
Die Ursache für die Entwicklung des Edwards-Syndroms ist die Verdreifachung des Chromosoms 18. Die Trisomie auf Chromosom 18 ist ein Sonderfall der Aneuploidie – das Vorhandensein eines Chromosomensatzes im Genom, der sich vom Standardsatz einer bestimmten Art unterscheidet und nicht ein Vielfaches davon. Trisomie 18 wird in der Regel durch eine Nichtdisjunktion der Chromosomen während der Bildung der Geschlechtszellen (Eizellen und Spermien) der Eltern verursacht, was dazu führt, dass das Kind ein zusätzliches 18. Chromosom von der Mutter oder dem Vater erhält. In diesem Fall tragen alle Zellen im Körper des Kindes die Anomalie. Wenn während der Teilung einer embryonalen Zelle eine Chromosomen-Nichtdisjunktion auftritt, wird eine Mosaikversion des Edwards-Syndroms beobachtet (10 % der Fälle).
Das Risiko, Kinder mit Edwards-Syndrom zu bekommen, verändert sich verschiedenen Literaturangaben zufolge mit zunehmendem Alter der Schwangeren nicht bzw. steigt leicht an.
Die pränatale Diagnose des Edwards-Syndroms umfasst zwei Stadien. Im ersten Stadium, in der 11. bis 13. Schwangerschaftswoche, wird ein Screening durchgeführt, das hauptsächlich auf biochemischen Indikatoren basiert, da der Ultraschall im Frühstadium beim Edwards-Syndrom keine groben Entwicklungsanomalien erkennt, was nur möglich ist nach 20–24 Wochen erkannt. Biochemische Analyse des Spiegels bestimmter Proteine im Blut einer schwangeren Frau (freie β-Untereinheit des menschlichen Chorionhormons (β-hCG) und schwangerschaftsassoziiertes Plasmaprotein A (PAPP-A)) unter Berücksichtigung ihres Alters, ermöglicht es uns zu berechnen, dass das Risiko besteht, ein krankes Kind zu bekommen. Diese Methoden ermöglichen jedoch keine genaue Diagnose und durch das Screening bildet sich eine Risikogruppe schwangerer Frauen mit einer erhöhten Wahrscheinlichkeit, eine Patientin mit Edwards-Syndrom zur Welt zu bringen. Im zweiten Schritt wird in der Risikogruppe ein invasiver Eingriff durchgeführt, um fetales Material zu gewinnen, das zur genauen Bestimmung des Status des Fötus erforderlich ist. Je nach Schwangerschaftsstadium kann es sich dabei um eine Chorionzottenbiopsie (8.–12. Woche), eine Amniozentese (14.–18. Woche) oder eine Cordozentese (nach der 20. Woche) handeln. In den gewonnenen fetalen Gewebeproben wird der Chromosomensatz bestimmt.
Das Zentrum für Molekulare Genetik diagnostiziert das Edwards-Syndrom (auch pränatal) mit der QF-PCR-Methode.
Edwards-Syndrom (Trisomie 18). Ursachen, Symptome, Anzeichen, Diagnose und Behandlung der Pathologie
Die Website bietet Hintergrundinformation. Unter Aufsicht eines gewissenhaften Arztes ist eine adäquate Diagnose und Behandlung der Erkrankung möglich.
 Edwards-Syndrom oder Trisomie 18 ist eine schwere angeborene Erkrankung, die durch Chromosomenanomalien verursacht wird. Es ist eine der häufigsten Pathologien in dieser Kategorie ( Zweithäufigste Erkrankung nach dem Down-Syndrom). Die Krankheit ist durch zahlreiche Störungen in der Entwicklung verschiedener Organe und Systeme gekennzeichnet. Die Prognose für das Kind ist meist ungünstig, doch hängt viel von der Betreuung durch die Eltern ab.
Edwards-Syndrom oder Trisomie 18 ist eine schwere angeborene Erkrankung, die durch Chromosomenanomalien verursacht wird. Es ist eine der häufigsten Pathologien in dieser Kategorie ( Zweithäufigste Erkrankung nach dem Down-Syndrom). Die Krankheit ist durch zahlreiche Störungen in der Entwicklung verschiedener Organe und Systeme gekennzeichnet. Die Prognose für das Kind ist meist ungünstig, doch hängt viel von der Betreuung durch die Eltern ab.
Die weltweite Prävalenz des Edwards-Syndroms schwankt zwischen 0,015 und 0,02 %. Es besteht keine eindeutige Abhängigkeit von Gebiet oder Rasse. Statistisch gesehen erkranken Mädchen drei- bis viermal häufiger als Jungen. Eine wissenschaftliche Erklärung für diesen Anteil konnte bislang nicht gefunden werden. Es wurde jedoch eine Reihe von Faktoren festgestellt, die das Risiko dieser Pathologie erhöhen können.
Wie andere Chromosomenmutationen ist das Edwards-Syndrom grundsätzlich eine unheilbare Krankheit. Am meisten moderne Methoden Behandlung und Pflege können das Kind nur am Leben erhalten und zu einem gewissen Fortschritt in seiner Entwicklung beitragen. Aufgrund der Vielzahl möglicher Erkrankungen und Komplikationen gibt es keine einheitlichen Empfehlungen für die Betreuung dieser Kinder.
Interessante Fakten
- Eine Beschreibung der Hauptsymptome dieser Krankheit erfolgte zu Beginn des 20. Jahrhunderts.
- Bis Mitte des 20. Jahrhunderts war es nicht möglich, ausreichende Informationen über diese Pathologie zu sammeln. Erstens erforderte dies eine entsprechende technologische Entwicklung, die den Nachweis des zusätzlichen Chromosoms ermöglichen würde. Zweitens starben die meisten Kinder bereits in den ersten Tagen oder Wochen ihres Lebens aufgrund der schlechten medizinischen Versorgung.
- Erste Gesamte Beschreibung Krankheit und ihre Hauptursache ( Auftreten eines zusätzlichen 18. Chromosoms) wurde erst 1960 von dem Arzt John Edward entwickelt, nach dem die neue Pathologie damals benannt wurde.
- Die tatsächliche Inzidenz des Edwards-Syndroms beträgt 1 Fall bei 2,5 bis 3.000 Empfängnissen ( 0,03 – 0,04% ), allerdings sind die offiziellen Daten viel niedriger. Dies erklärt sich dadurch, dass fast die Hälfte der Embryonen mit dieser Anomalie nicht überleben und die Schwangerschaft mit einem Spontanabort oder dem intrauterinen Tod des Fötus endet. Eine detaillierte Diagnose der Ursache einer Fehlgeburt wird selten durchgeführt.
- Trisomie ist eine Variante einer Chromosomenmutation, bei der die Zellen einer Person nicht 46, sondern 47 Chromosomen enthalten. In dieser Krankheitsgruppe gibt es nur 3 Syndrome. Neben dem Edwards-Syndrom sind dies Down-Syndrome ( Trisomie 21 Chromosom) und Patau ( Trisomie 13 Chromosom). Bei Vorhandensein weiterer zusätzlicher Chromosomen ist die Pathologie nicht mit dem Leben vereinbar. Nur in diesen drei Fällen ist die Geburt eines lebenden Kindes und dessen weitere ( wenn auch langsam) Wachstum und Entwicklung.
Ursachen der genetischen Pathologie
 Edwards-Syndrom ist Erbkrankheit, die durch das Vorhandensein eines zusätzlichen Chromosoms im menschlichen Genom gekennzeichnet ist. Um die Gründe zu verstehen, die die sichtbaren Manifestationen dieser Pathologie verursachen, ist es notwendig herauszufinden, was die Chromosomen selbst und das genetische Material als Ganzes sind.
Edwards-Syndrom ist Erbkrankheit, die durch das Vorhandensein eines zusätzlichen Chromosoms im menschlichen Genom gekennzeichnet ist. Um die Gründe zu verstehen, die die sichtbaren Manifestationen dieser Pathologie verursachen, ist es notwendig herauszufinden, was die Chromosomen selbst und das genetische Material als Ganzes sind.
Jede menschliche Zelle verfügt über einen Zellkern, der für die Speicherung und Verarbeitung genetischer Informationen verantwortlich ist. Der Kern enthält 46 Chromosomen ( 23 Paare), bei denen es sich um ein mehrfach verpacktes DNA-Molekül handelt ( Desoxyribonukleinsäure). Dieses Molekül enthält bestimmte Abschnitte, die Gene genannt werden. Jedes Gen ist der Prototyp eines bestimmten Proteins im menschlichen Körper. Bei Bedarf liest die Zelle Informationen aus diesem Prototyp aus und produziert das entsprechende Protein. Gendefekte führen zur Produktion abnormaler Proteine, die für die Entstehung genetischer Krankheiten verantwortlich sind.
Ein Chromosomenpaar besteht aus zwei identischen DNA-Molekülen ( das eine ist väterlicherseits, das andere mütterlicherseits), die durch eine kleine Brücke miteinander verbunden sind ( Zentromer). Der Ort der Verbindung zweier Chromosomen eines Paares bestimmt die Form der gesamten Verbindung und ihr Aussehen unter dem Mikroskop.
Alle Chromosomen speichern unterschiedliche genetische Informationen (über verschiedene Proteine) und werden in folgende Gruppen eingeteilt:
- Gruppe A enthält 1 – 3 Chromosomenpaare, die groß und X-förmig sind;
- Gruppe B enthält 4–5 Chromosomenpaare, die ebenfalls groß sind, aber das Zentromer liegt weiter vom Zentrum entfernt, weshalb die Form dem Buchstaben X ähnelt, wobei das Zentrum nach unten oder oben verschoben ist;
- Gruppe C umfasst 6–12 Chromosomenpaare, die in ihrer Form den Chromosomen der Gruppe B ähneln, ihnen jedoch in der Größe unterlegen sind;
- Gruppe D umfasst 13 - 15 Chromosomenpaare, die sich durch mittlere Größe und die Lage des Zentromers ganz am Ende der Moleküle auszeichnen, was ihm eine Ähnlichkeit mit dem Buchstaben V verleiht;
- Gruppe E umfasst 16–18 Chromosomenpaare, die durch geringe Größe und mittlere Lage des Zentromers gekennzeichnet sind ( Buchstabe X-Form);
- Gruppe F umfasst 19–20 Chromosomenpaare, die etwas kleiner als Chromosomen der Gruppe E und ähnlich geformt sind;
- Gruppe G umfasst 21–22 Chromosomenpaare, die sich durch eine V-Form und eine sehr geringe Größe auszeichnen.
Die oben genannten 22 Chromosomenpaare werden somatisch oder Autosomen genannt. Darüber hinaus gibt es Geschlechtschromosomen, die das 23. Paar bilden. Da sie sich im Aussehen nicht ähneln, wird jeder von ihnen separat bezeichnet. Das weibliche Geschlechtschromosom wird mit X bezeichnet und ähnelt der Gruppe C. Das männliche Geschlechtschromosom wird mit Y bezeichnet und ähnelt in Form und Größe der Gruppe G. Wenn ein Kind beide Chromosomen weiblich hat ( Typ XX), dann wird ein Mädchen geboren. Wenn eines der Geschlechtschromosomen weiblich und das andere männlich ist, wird ein Junge geboren ( Typ XY). Die Chromosomenformel wird als Karyotyp bezeichnet und kann bezeichnet werden auf die folgende Weise— 46, XX. Hier bezeichnet die Zahl 46 die Gesamtzahl der Chromosomen ( 23 Paare) und XX ist die Formel der Geschlechtschromosomen, die vom Geschlecht abhängt ( Das Beispiel zeigt den Karyotyp einer normalen Frau).
Unter dem Edwards-Syndrom versteht man sogenannte Chromosomenerkrankungen, bei denen es sich nicht um einen Gendefekt, sondern um einen Defekt im gesamten DNA-Molekül handelt. Genauer gesagt handelt es sich bei der klassischen Form dieser Krankheit um das Vorhandensein eines zusätzlichen 18. Chromosoms. Der Karyotyp wird in solchen Fällen als 47,XX, 18+ ( für Mädchen) und 47,ХY, 18+ ( für Junge). Die letzte Ziffer gibt die Nummer des zusätzlichen Chromosoms an. Überschüssige genetische Informationen in Zellen führen zum Auftreten entsprechender Krankheitserscheinungen, die zusammenfassend als „Edwards-Syndrom“ bezeichnet werden. Verfügbarkeit zusätzlicher ( dritte) Chromosom Nummer 18 ergab ein weiteres ( wissenschaftlicher) Die Krankheit heißt Trisomie 18.
Abhängig von der Form des Chromosomendefekts werden drei Arten dieser Erkrankung unterschieden:
- Vollständige Trisomie 18. Bei der vollständigen oder klassischen Form des Edwards-Syndroms verfügen alle Zellen im Körper über ein zusätzliches Chromosom. Diese Option Die Erkrankung tritt in mehr als 90 % der Fälle auf und verläuft am schwersten.
- Partielle Trisomie 18. Partielle Trisomie 18 ist ein sehr seltenes Phänomen ( nicht mehr als 3 % aller Fälle von Edwards-Syndrom). Damit enthalten die Körperzellen nicht ein ganzes zusätzliches Chromosom, sondern nur einen Bruchteil davon. Dieser Defekt kann die Folge einer unsachgemäßen Aufteilung des genetischen Materials sein, kommt aber sehr selten vor. Manchmal ist ein Teil des achtzehnten Chromosoms an ein anderes DNA-Molekül gebunden ( wird in seine Struktur eingeführt, verlängert das Molekül oder „klebt“ einfach mit Hilfe einer Brücke). Die anschließende Zellteilung führt dazu, dass der Körper über 2 normale Chromosomen Nummer 18 und einige weitere Gene dieser Chromosomen verfügt ( konserviertes Fragment eines DNA-Moleküls). In diesem Fall die Menge Geburtsfehler wird viel niedriger sein. Es gibt nicht die gesamte im 18. Chromosom kodierte genetische Information, sondern nur einen Teil davon. Patienten mit partieller Trisomie 18 haben eine bessere Prognose als Kinder mit der Vollform, bleiben aber weiterhin schlecht.
- Mosaikform. Die Mosaikform des Edwards-Syndroms tritt in 5–7 % der Fälle dieser Erkrankung auf. Der Mechanismus seines Auftretens unterscheidet sich von anderen Arten. Tatsache ist, dass hier der Defekt nach der Verschmelzung von Sperma und Ei entstanden ist. Beide Gameten ( Keimzellen) hatte ursprünglich einen normalen Karyotyp und trug ein Chromosom jeder Art. Nach der Fusion entstand eine Zelle mit der normalen Formel 46,XX oder 46,XY. Bei der Teilung dieser Zelle ist eine Fehlfunktion aufgetreten. Bei der Verdoppelung des genetischen Materials erhielt eines der Fragmente ein zusätzliches 18. Chromosom. So hat sich zu einem bestimmten Zeitpunkt ein Embryo gebildet, dessen Zellen teilweise einen normalen Karyotyp aufweisen ( zum Beispiel 46,XX), und ein Teil ist ein Karyotyp des Edwards-Syndroms ( 47,XX, 18+). Der Anteil pathologischer Zellen übersteigt nie 50 %. Ihre Anzahl hängt davon ab, in welchem Teilungsstadium der Ausgangszelle der Fehler aufgetreten ist. Je später dies geschieht, desto geringer ist der Anteil defekter Zellen. Die Form erhielt ihren Namen aufgrund der Tatsache, dass alle Zellen des Körpers eine Art Mosaik darstellen. Einige von ihnen sind gesund, andere haben eine schwere genetische Pathologie. In diesem Fall gibt es kein Muster in der Verteilung der Zellen im Körper, das heißt, alle defekten Zellen können nicht an nur einer Stelle lokalisiert werden, um sie entfernen zu können. Der Allgemeinzustand des Patienten ist einfacher als bei der klassischen Form der Trisomie 18.
Das Vorhandensein eines zusätzlichen Chromosoms im menschlichen Genom bringt viele Probleme mit sich. Tatsache ist, dass menschliche Zellen darauf programmiert sind, genetische Informationen zu lesen und nur die von der Natur vorgegebene Anzahl an DNA-Molekülen zu duplizieren. Störungen bereits in der Struktur eines Gens können zu schweren Krankheiten führen. Bei Vorhandensein eines vollständigen DNA-Moleküls kommt es bereits im Stadium der intrauterinen Entwicklung vor der Geburt des Kindes zu multiplen Störungen.
Jüngsten Forschungsergebnissen zufolge enthält das Chromosom Nummer 18 557 Gene, die für mindestens 289 verschiedene Proteine kodieren. Prozentual sind das etwa 2,5 % des gesamten Erbguts. Die Störungen, die ein so großes Ungleichgewicht verursacht, sind sehr schwerwiegend. Eine falsche Menge an Proteinen führt zu vielen Anomalien in der Entwicklung verschiedener Organe und Gewebe. Beim Edwards-Syndrom sind am häufigsten die Schädelknochen, einige Teile des Nervensystems, das Herz-Kreislauf- und Urogenitalsystem betroffen. Dies liegt offenbar daran, dass die auf diesem Chromosom befindlichen Gene mit der Entwicklung dieser bestimmten Organe und Systeme zusammenhängen.
Somit ist die Haupt- und einzige Ursache des Edwards-Syndroms das Vorhandensein eines zusätzlichen DNA-Moleküls. Meistens ( in der klassischen Form der Erkrankung) wird von einem der Eltern geerbt. Normalerweise ist jeder Gamete ( Sperma und Ei) enthalten 22 ungepaarte somatische Chromosomen sowie ein Geschlechtschromosom. Eine Frau gibt dem Kind immer den Standardsatz 22+X weiter, und ein Mann kann 22+X oder 22+Y weitergeben. Dadurch wird das Geschlecht des Kindes bestimmt. Die Geschlechtszellen der Eltern entstehen durch die Teilung gewöhnlicher Zellen in zwei Gruppen. Normalerweise ist die Mutterzelle in zwei gleiche Teile geteilt, manchmal sind jedoch nicht alle Chromosomen in zwei Hälften geteilt. Wenn sich das 18. Paar nicht an den Polen der Zelle getrennt hat, dann ist eines der Eier ( oder eines der Spermien) wird von vornherein defekt sein. Es wird nicht 23, sondern 24 Chromosomen haben. Wenn diese Zelle an der Befruchtung beteiligt ist, erhält das Kind ein zusätzliches 18. Chromosom.
Die folgenden Faktoren können eine fehlerhafte Zellteilung beeinflussen:
- Alter der Eltern. Es ist erwiesen, dass die Wahrscheinlichkeit von Chromosomenanomalien direkt proportional zum Alter der Mutter steigt. Beim Edwards-Syndrom ist dieser Zusammenhang weniger ausgeprägt als bei anderen ähnlichen Pathologien ( zum Beispiel Down-Syndrom). Bei Frauen über 40 Jahren ist das Risiko, ein Kind mit dieser Pathologie zu bekommen, jedoch im Durchschnitt sechs- bis siebenmal höher. Diese Abhängigkeit vom Alter des Vaters ist in deutlich geringerem Maße zu beobachten.
- Rauchen und Alkohol. Schlechte Gewohnheiten wie Rauchen und Alkoholmissbrauch können das menschliche Fortpflanzungssystem beeinträchtigen und die Teilung der Keimzellen beeinträchtigen. Daher ist die regelmäßige Einnahme dieser Substanzen ( sowie andere Betäubungsmittel) erhöht das Risiko einer unsachgemäßen Verteilung von genetischem Material.
- Rezeption Medikamente . Manche Medikamente Bei falscher Einnahme im ersten Trimester können sie die Teilung der Keimzellen beeinträchtigen und die Mosaikform des Edwards-Syndroms hervorrufen.
- Erkrankungen des Genitalbereichs. Frühere Infektionen der Fortpflanzungsorgane können die ordnungsgemäße Zellteilung beeinträchtigen. Sie erhöhen das Risiko für chromosomale und genetische Erkrankungen im Allgemeinen, obwohl ähnliche Studien speziell für das Edwards-Syndrom nicht durchgeführt wurden.
- Strahlung. Die Einwirkung von Röntgenstrahlen oder anderer ionisierender Strahlung auf die Genitalien kann zu genetischen Mutationen führen. Besonders gefährlich sind solche äußeren Einflüsse im Jugendalter, wenn die Zellteilung am aktivsten erfolgt. Die Partikel, aus denen die Strahlung besteht, dringen leicht in das Gewebe ein und setzen das DNA-Molekül einer Art „Bombardierung“ aus. Geschieht dies zum Zeitpunkt der Zellteilung, ist das Risiko einer Chromosomenmutation besonders hoch.
Im Allgemeinen kann man nicht sagen, dass die Ursachen für die Entstehung des Edwards-Syndroms vollständig bekannt und gut untersucht sind. Die oben genannten Faktoren erhöhen nur das Risiko, diese Mutation zu entwickeln. Es ist auch möglich, dass manche Menschen eine angeborene Veranlagung für eine falsche Verteilung des genetischen Materials in den Keimzellen haben. Man geht beispielsweise davon aus, dass ein Ehepaar, das bereits ein Kind mit Edwards-Syndrom zur Welt gebracht hat, eine Wahrscheinlichkeit von 2–3 % hat, ein zweites Kind mit einer ähnlichen Pathologie zu bekommen ( etwa 200-mal höher als die durchschnittliche Prävalenz der Krankheit).
Wie sehen Neugeborene mit Edwards-Syndrom aus?
 Wie Sie wissen, kann das Edwards-Syndrom bereits vor der Geburt diagnostiziert werden, in den meisten Fällen wird diese Krankheit jedoch unmittelbar nach der Geburt des Kindes erkannt. Neugeborene mit dieser Pathologie weisen eine Reihe ausgeprägter Entwicklungsanomalien auf, die manchmal sofort den Verdacht auf die richtige Diagnose zulassen. Die Bestätigung erfolgt später durch eine spezielle genetische Analyse.
Wie Sie wissen, kann das Edwards-Syndrom bereits vor der Geburt diagnostiziert werden, in den meisten Fällen wird diese Krankheit jedoch unmittelbar nach der Geburt des Kindes erkannt. Neugeborene mit dieser Pathologie weisen eine Reihe ausgeprägter Entwicklungsanomalien auf, die manchmal sofort den Verdacht auf die richtige Diagnose zulassen. Die Bestätigung erfolgt später durch eine spezielle genetische Analyse.
Neugeborene mit Edwards-Syndrom weisen folgende charakteristische Entwicklungsstörungen auf:
- Veränderung der Schädelform;
- Veränderung der Ohrenform;
- Anomalien der Gaumenentwicklung;
- Wippenfuß;
- abnormale Fingerlänge;
- Veränderung der Form des Unterkiefers;
- Verschmelzung der Finger;
- abnormale Entwicklung der Geschlechtsorgane;
- Beugestellung der Hände;
- dermatoglyphische Zeichen.
Die Form des Schädels verändern
 Ein typisches Symptom des Edwards-Syndroms ist die Dolichozephalie. Dies ist die Bezeichnung für eine charakteristische Veränderung der Kopfform eines Neugeborenen, die auch bei einigen anderen genetisch bedingten Erkrankungen auftritt. Bei Dolichocephalen ( Kinder mit diesem Symptom) längerer und schmalerer Schädel. Das Vorliegen dieser Anomalie wird durch spezielle Messungen genau bestätigt. Bestimmen Sie das Verhältnis der Schädelbreite auf Höhe der Scheitelknochen zur Schädellänge ( vom Vorsprung über dem Nasenrücken bis zum Hinterhauptsvorsprung). Wenn das resultierende Verhältnis weniger als 75 % beträgt, dann dieses Kind bezieht sich auf Dolichocephals. Dieses Symptom an sich ist keine ernsthafte Störung. Dies ist einfach eine der Arten von Schädelformen, die absolut vorkommen normale Leute. Bei Kindern mit Edwards-Syndrom handelt es sich in 80 - 85 % der Fälle um ausgeprägte Dolichocephalen, bei denen ohne besondere Messungen das Missverhältnis von Schädellänge und -breite auffällt.
Ein typisches Symptom des Edwards-Syndroms ist die Dolichozephalie. Dies ist die Bezeichnung für eine charakteristische Veränderung der Kopfform eines Neugeborenen, die auch bei einigen anderen genetisch bedingten Erkrankungen auftritt. Bei Dolichocephalen ( Kinder mit diesem Symptom) längerer und schmalerer Schädel. Das Vorliegen dieser Anomalie wird durch spezielle Messungen genau bestätigt. Bestimmen Sie das Verhältnis der Schädelbreite auf Höhe der Scheitelknochen zur Schädellänge ( vom Vorsprung über dem Nasenrücken bis zum Hinterhauptsvorsprung). Wenn das resultierende Verhältnis weniger als 75 % beträgt, dann dieses Kind bezieht sich auf Dolichocephals. Dieses Symptom an sich ist keine ernsthafte Störung. Dies ist einfach eine der Arten von Schädelformen, die absolut vorkommen normale Leute. Bei Kindern mit Edwards-Syndrom handelt es sich in 80 - 85 % der Fälle um ausgeprägte Dolichocephalen, bei denen ohne besondere Messungen das Missverhältnis von Schädellänge und -breite auffällt.
Eine weitere Variante der Fehlentwicklung des Schädels ist die sogenannte Mikrozephalie, bei der der Kopf insgesamt im Vergleich zum Rest des Körpers zu klein ist. Dies gilt zunächst nicht für den Gesichtsschädel ( Kiefer, Wangenknochen, Augenhöhlen), nämlich der Schädel, in dem sich das Gehirn befindet. Mikrozephalie ist beim Edwards-Syndrom seltener als Dolichozephalie, kommt aber auch häufiger vor als bei anderen gesunde Menschen.
Die Form des Ohrs verändern
 Wenn Dolichozephalie eine normale Variante sein kann, ist die Pathologie der Entwicklung der Ohrmuschel bei Kindern mit Edwards-Syndrom viel schwerwiegender. Bis zu einem gewissen Grad wird dieses Symptom bei mehr als 95 % der Kinder mit der Vollform dieser Krankheit beobachtet. In Mosaikform ist seine Häufigkeit etwas geringer. Die Ohrmuschel liegt normalerweise tiefer als bei normalen Menschen ( manchmal unter Augenhöhe). Die charakteristischen Höcker des Knorpels, der die Ohrmuschel bildet, sind schlecht definiert oder fehlen. Der Lappen oder Tragus kann auch fehlen ( ein kleiner hervorstehender Knorpelbereich vor der Gehöröffnung). Der Gehörgang selbst ist meist verengt und fehlt in etwa 20–25 % vollständig.
Wenn Dolichozephalie eine normale Variante sein kann, ist die Pathologie der Entwicklung der Ohrmuschel bei Kindern mit Edwards-Syndrom viel schwerwiegender. Bis zu einem gewissen Grad wird dieses Symptom bei mehr als 95 % der Kinder mit der Vollform dieser Krankheit beobachtet. In Mosaikform ist seine Häufigkeit etwas geringer. Die Ohrmuschel liegt normalerweise tiefer als bei normalen Menschen ( manchmal unter Augenhöhe). Die charakteristischen Höcker des Knorpels, der die Ohrmuschel bildet, sind schlecht definiert oder fehlen. Der Lappen oder Tragus kann auch fehlen ( ein kleiner hervorstehender Knorpelbereich vor der Gehöröffnung). Der Gehörgang selbst ist meist verengt und fehlt in etwa 20–25 % vollständig.
Anomalien der Gaumenentwicklung
 Die Gaumenfortsätze des Oberkiefers verschmelzen während der Embryonalentwicklung zum harten Gaumen. Bei Kindern mit Edwards-Syndrom bleibt dieser Prozess oft unvollständig. An der Stelle, an der sich bei normalen Menschen die Mittelnaht befindet ( man kann es mit der Zunge in der Mitte des harten Gaumens ertasten) Sie haben einen Längsschlitz.
Die Gaumenfortsätze des Oberkiefers verschmelzen während der Embryonalentwicklung zum harten Gaumen. Bei Kindern mit Edwards-Syndrom bleibt dieser Prozess oft unvollständig. An der Stelle, an der sich bei normalen Menschen die Mittelnaht befindet ( man kann es mit der Zunge in der Mitte des harten Gaumens ertasten) Sie haben einen Längsschlitz.
Es gibt mehrere Varianten dieses Defekts:
- Gaumensegelspalte ( der hintere, tiefe Teil des Gaumens, der über der Kehle hängt);
- teilweise Verschmelzung des harten Gaumens ( Die Lücke erstreckt sich nicht über den gesamten Oberkiefer);
- völlige Nichtverschmelzung des harten und weichen Gaumens;
- völlige Nichtverschmelzung von Gaumen und Lippe.
In einigen Fällen ist die Gaumenspalte beidseitig. Die beiden nach oben ragenden Ecken der Oberlippe sind der Beginn pathologischer Risse. Aufgrund dieses Defekts kann das Kind seinen Mund nicht vollständig schließen. In schweren Fällen ist die Kommunikation zwischen Mund- und Nasenhöhle deutlich sichtbar ( auch mit geschlossenem Mund). Vorderzähne können in Zukunft fehlen oder zur Seite wachsen.
Diese Entwicklungsstörungen werden auch als Gaumenspalte, Gaumenspalte und Lippenspalte bezeichnet. Alle von ihnen können außerhalb des Edwards-Syndroms auftreten, aber bei Kindern mit dieser Pathologie ist ihre Häufigkeit besonders hoch ( fast 20 % der Neugeborenen). Viel häufiger ( bis zu 65 % der Neugeborenen) haben ein weiteres Merkmal, das als hoher oder gotischer Himmel bekannt ist. Sie kann als normale Variante eingestuft werden, da sie auch bei gesunden Menschen auftritt.
Das Vorhandensein einer Gaumen- oder Oberlippenspalte ist kein Beweis für das Edwards-Syndrom. Diese Fehlbildung kann mit relativ hoher Häufigkeit und unabhängig voneinander ohne Begleitstörungen anderer Organe und Systeme auftreten. Um diese Anomalie zu korrigieren, gibt es eine Reihe standardmäßiger chirurgischer Eingriffe.
Schaukelfuß
 So bezeichnet man eine charakteristische Fußveränderung, die vor allem im Rahmen des Edwards-Syndroms auftritt. Die Häufigkeit bei dieser Krankheit beträgt 75 %. Der Defekt besteht in einer falschen Beziehung zwischen Talus, Fersenbein und Strahlbein. Bei Kindern wird sie als Plattvalgusfußdeformität klassifiziert.
So bezeichnet man eine charakteristische Fußveränderung, die vor allem im Rahmen des Edwards-Syndroms auftritt. Die Häufigkeit bei dieser Krankheit beträgt 75 %. Der Defekt besteht in einer falschen Beziehung zwischen Talus, Fersenbein und Strahlbein. Bei Kindern wird sie als Plattvalgusfußdeformität klassifiziert.
Äußerlich sieht der Fuß eines Neugeborenen so aus. Der Fersenhöcker, auf dem der Fußrücken aufliegt, ragt nach hinten. Der Bogen kann vollständig fehlen. Dies ist leicht zu erkennen, wenn man den Fuß von innen betrachtet. Normalerweise erscheint dort eine konkave Linie, die von der Ferse bis zur Basis des großen Zehs verläuft. Bei einem Kippfuß gibt es diese Linie nicht. Der Fuß ist flach oder sogar konvex. Dadurch sieht es aus wie die Beine eines Schaukelstuhls.
Abnormale Fingerlänge
 Kinder mit Edwards-Syndrom können aufgrund von Veränderungen in der Struktur ihrer Füße abnormale Längenproportionen ihrer Zehen aufweisen. Dabei handelt es sich insbesondere um den Daumen, der normalerweise der längste ist. Bei Neugeborenen mit diesem Syndrom ist die Länge geringer als der zweite Finger. Dieser Defekt kann nur bemerkt werden, wenn die Finger gestreckt und sorgfältig untersucht werden. Mit zunehmendem Alter, wenn das Kind wächst, wird es deutlicher. Da die Verkürzung des Hallux vor allem bei Kippfüßen auftritt, ist die Prävalenz dieser Symptome bei Neugeborenen ungefähr gleich.
Kinder mit Edwards-Syndrom können aufgrund von Veränderungen in der Struktur ihrer Füße abnormale Längenproportionen ihrer Zehen aufweisen. Dabei handelt es sich insbesondere um den Daumen, der normalerweise der längste ist. Bei Neugeborenen mit diesem Syndrom ist die Länge geringer als der zweite Finger. Dieser Defekt kann nur bemerkt werden, wenn die Finger gestreckt und sorgfältig untersucht werden. Mit zunehmendem Alter, wenn das Kind wächst, wird es deutlicher. Da die Verkürzung des Hallux vor allem bei Kippfüßen auftritt, ist die Prävalenz dieser Symptome bei Neugeborenen ungefähr gleich.
Bei Erwachsenen hat eine Verkürzung der Großzehe nicht den gleichen diagnostischen Wert. Ein solcher Defekt kann ein individuelles Merkmal eines gesunden Menschen oder eine Folge des Einflusses anderer Faktoren sein ( Verformung der Gelenke, Knochenerkrankungen, Tragen von Schuhen, die nicht richtig passen). In diesem Zusammenhang sollte dieses Zeichen nur bei Neugeborenen als mögliches Symptom in Betracht gezogen werden, wenn andere Entwicklungsanomalien vorliegen.
Veränderung der Form des Unterkiefers
 Formveränderungen des Unterkiefers treten bei Neugeborenen in fast 70 % der Fälle auf. Normalerweise ragt das Kinn bei Kindern nicht so stark hervor wie bei Erwachsenen, bei Patienten mit Edwards-Syndrom ist es jedoch zu stark zurückgezogen. Dies geschieht aufgrund einer Unterentwicklung des Unterkiefers, die als Mikrognathie bezeichnet wird ( Mikrogenie). Dieses Symptom tritt auch bei anderen angeborenen Erkrankungen auf. Es ist nicht so selten, Erwachsene mit ähnlichen Gesichtszügen zu finden. In Ermangelung begleitender Pathologien gilt dies als Variante der Norm, führt jedoch zu einigen Schwierigkeiten.
Formveränderungen des Unterkiefers treten bei Neugeborenen in fast 70 % der Fälle auf. Normalerweise ragt das Kinn bei Kindern nicht so stark hervor wie bei Erwachsenen, bei Patienten mit Edwards-Syndrom ist es jedoch zu stark zurückgezogen. Dies geschieht aufgrund einer Unterentwicklung des Unterkiefers, die als Mikrognathie bezeichnet wird ( Mikrogenie). Dieses Symptom tritt auch bei anderen angeborenen Erkrankungen auf. Es ist nicht so selten, Erwachsene mit ähnlichen Gesichtszügen zu finden. In Ermangelung begleitender Pathologien gilt dies als Variante der Norm, führt jedoch zu einigen Schwierigkeiten.
Neugeborene mit Mikrognathie entwickeln in der Regel schnell folgende Probleme:
- Unfähigkeit, den Mund lange geschlossen zu halten ( Austreten von Speichel);
- Schwierigkeiten beim Füttern;
- späte Entwicklung der Zähne und deren falsche Lage.
Die Lücke zwischen unten und Oberkiefer kann mehr als 1 cm betragen, was angesichts der Kopfgröße des Babys viel ist.
Fingerfusion
 Bei etwa 45 % der Neugeborenen wird eine Verschmelzung der Finger oder wissenschaftlich Syndaktylie beobachtet. Am häufigsten betrifft diese Anomalie die Zehen, Syndaktylie tritt jedoch auch an den Händen auf. In milden Fällen wird die Fusion durch eine Hautfalte wie eine kurze Membran gebildet. In schwereren Fällen wird eine Fusion von Knochengewebe durch Brücken beobachtet.
Bei etwa 45 % der Neugeborenen wird eine Verschmelzung der Finger oder wissenschaftlich Syndaktylie beobachtet. Am häufigsten betrifft diese Anomalie die Zehen, Syndaktylie tritt jedoch auch an den Händen auf. In milden Fällen wird die Fusion durch eine Hautfalte wie eine kurze Membran gebildet. In schwereren Fällen wird eine Fusion von Knochengewebe durch Brücken beobachtet.
Syndaktylie kommt nicht nur beim Edwards-Syndrom vor, sondern auch bei vielen anderen Chromosomenerkrankungen. Es gibt auch Fälle, in denen dieser Entwicklungsfehler der einzige war und sich der Patient ansonsten nicht von normalen Kindern unterschied. In dieser Hinsicht ist die Fingerfusion nur eines der möglichen Anzeichen des Edwards-Syndroms, das zur Vermutung der Diagnose beiträgt, diese jedoch nicht bestätigt.
Anomalien in der Entwicklung der Geschlechtsorgane
Beugehaltung der Hände
 Bei der Beugestellung der Hände handelt es sich um eine besondere Anordnung der Finger, die weniger auf Strukturstörungen im Handbereich als vielmehr auf einen erhöhten Muskeltonus zurückzuführen ist. Die Beugemuskeln der Finger und der Hand sind ständig angespannt, weshalb Daumen und kleiner Finger scheinbar die anderen Finger bedecken, die gegen die Handfläche gedrückt werden. Dieses Symptom wird bei vielen angeborenen Pathologien beobachtet und ist nicht spezifisch für das Edwards-Syndrom. Wenn jedoch ein Pinsel dieser Form entdeckt wird, muss von dieser Pathologie ausgegangen werden. Damit wird bei fast 90 % der Neugeborenen eine Beugestellung der Finger beobachtet.
Bei der Beugestellung der Hände handelt es sich um eine besondere Anordnung der Finger, die weniger auf Strukturstörungen im Handbereich als vielmehr auf einen erhöhten Muskeltonus zurückzuführen ist. Die Beugemuskeln der Finger und der Hand sind ständig angespannt, weshalb Daumen und kleiner Finger scheinbar die anderen Finger bedecken, die gegen die Handfläche gedrückt werden. Dieses Symptom wird bei vielen angeborenen Pathologien beobachtet und ist nicht spezifisch für das Edwards-Syndrom. Wenn jedoch ein Pinsel dieser Form entdeckt wird, muss von dieser Pathologie ausgegangen werden. Damit wird bei fast 90 % der Neugeborenen eine Beugestellung der Finger beobachtet.
Dermatoglyphische Zeichen
 Viele Chromosomenanomalien bei Neugeborenen weisen charakteristische dermatoglyphische Veränderungen auf ( abnormale Muster und Falten auf der Haut der Handflächen). Beim Edwards-Syndrom können in fast 60 % der Fälle einige Anzeichen festgestellt werden. Sie sind vor allem für die vorläufige Diagnose von Mosaik- oder Teilformen der Erkrankung wichtig. Bei vollständiger Trisomie 18 wird nicht auf Dermatoglyphen zurückgegriffen, da andere, auffälligere Entwicklungsanomalien ausreichen, um ein Edwards-Syndrom zu vermuten.
Viele Chromosomenanomalien bei Neugeborenen weisen charakteristische dermatoglyphische Veränderungen auf ( abnormale Muster und Falten auf der Haut der Handflächen). Beim Edwards-Syndrom können in fast 60 % der Fälle einige Anzeichen festgestellt werden. Sie sind vor allem für die vorläufige Diagnose von Mosaik- oder Teilformen der Erkrankung wichtig. Bei vollständiger Trisomie 18 wird nicht auf Dermatoglyphen zurückgegriffen, da andere, auffälligere Entwicklungsanomalien ausreichen, um ein Edwards-Syndrom zu vermuten.
Die wichtigsten dermatoglyphischen Zeichen des Edwards-Syndroms sind:
- Bögen an den Fingerspitzen sind häufiger zu finden als bei gesunden Menschen;
- Hautfalte zwischen der letzten ( Nagel) und vorletzte ( Median) die Fingerglieder fehlen;
- 30 % der Neugeborenen haben eine sogenannte Querfurche an der Handfläche ( Affenlinie, Affenlinie).
Spezielle Untersuchungen können andere Abweichungen von der Norm aufdecken, aber unmittelbar nach der Geburt, ohne Einbeziehung spezialisierter Spezialisten, reichen diese Veränderungen für Ärzte aus.
Zusätzlich zu den oben genannten Anzeichen gibt es eine Reihe möglicher Entwicklungsanomalien, die bei der vorläufigen Diagnose des Edwards-Syndroms hilfreich sein können. Einigen Daten zufolge kann eine detaillierte äußere Untersuchung bis zu 50 äußere Anzeichen erkennen. Die Kombination der meisten häufige Symptome Die oben dargestellten Ergebnisse weisen mit hoher Wahrscheinlichkeit auf das Vorliegen dieser schweren Pathologie beim Kind hin. Bei der mosaikartigen Variante des Edwards-Syndroms kann es zwar nicht zu mehreren Anomalien kommen, aber schon das Vorliegen einer davon ist ein Hinweis auf einen speziellen Gentest.
Wie sehen Kinder mit Edwards-Syndrom aus?
 Kinder mit Edwards-Syndrom entwickeln mit zunehmendem Alter typischerweise eine Vielzahl von Begleiterkrankungen. Ihre Symptome treten bereits wenige Wochen nach der Geburt auf. Diese Symptome können die erste Manifestation des Syndroms sein, da bei der Mosaikvariante in seltenen Fällen die Krankheit unmittelbar nach der Geburt unbemerkt bleiben kann. Dann wird die Diagnose der Krankheit komplizierter.
Kinder mit Edwards-Syndrom entwickeln mit zunehmendem Alter typischerweise eine Vielzahl von Begleiterkrankungen. Ihre Symptome treten bereits wenige Wochen nach der Geburt auf. Diese Symptome können die erste Manifestation des Syndroms sein, da bei der Mosaikvariante in seltenen Fällen die Krankheit unmittelbar nach der Geburt unbemerkt bleiben kann. Dann wird die Diagnose der Krankheit komplizierter.
Die meisten der bei der Geburt festgestellten äußeren Erscheinungsformen des Syndroms bleiben bestehen und machen sich deutlicher bemerkbar. Wir sprechen über die Form des Schädels, den Kippfuß, die Verformung der Ohrmuschel usw. Allmählich kommen weitere äußere Erscheinungen hinzu, die unmittelbar nach der Geburt nicht bemerkt werden konnten. In diesem Fall handelt es sich um Anzeichen, die bei Kindern im ersten Lebensjahr auftreten können.
Kinder mit Edwards-Syndrom weisen folgende äußere Merkmale auf:
- zurückbleiben körperliche Entwicklung;
- Klumpfuß;
- abnormaler Muskeltonus;
- abnormale emotionale Reaktionen.
Verzögerte körperliche Entwicklung
Die Verzögerung der körperlichen Entwicklung erklärt sich durch das niedrige Geburtsgewicht des Kindes ( nur 2000 – 2200 g während einer normalen Schwangerschaft). Eine wesentliche Rolle spielt auch ein genetischer Defekt, der nicht dazu führt, dass sich alle Körpersysteme normal und harmonisch entwickeln können. Die Hauptindikatoren, anhand derer das Wachstum und die Entwicklung eines Kindes beurteilt werden, sind stark reduziert.
Sie können die Verzögerung eines Kindes anhand der folgenden anthropometrischen Indikatoren erkennen:
- Größe des Kindes;
- Gewicht des Kindes;
- Brustumfang;
- Kopfumfang ( Dieser Indikator kann normal oder sogar erhöht sein, ist jedoch aufgrund einer angeborenen Verformung des Schädels nicht verlässlich).
Klumpfuß
Anormaler Muskeltonus
Abnormale emotionale Reaktionen
Wie sehen Erwachsene mit Edwards-Syndrom aus?
 In den allermeisten Fällen erreichen Kinder, die mit dem Edwards-Syndrom geboren werden, nicht das Erwachsenenalter. In der Vollform dieser Krankheit, wenn in jeder Körperzelle ein zusätzliches Chromosom vorhanden ist, sterben 90 % der Kinder vor dem Alter von einem Jahr aufgrund schwerwiegender Anomalien in der Entwicklung innerer Organe. Auch bei chirurgischer Korrektur möglicher Defekte und hochwertiger Pflege ist ihr Körper anfälliger für Infektionskrankheiten. Begünstigt wird dies auch durch Essstörungen, die bei den meisten Kindern auftreten. All dies erklärt die höchste Sterblichkeitsrate beim Edwards-Syndrom.
In den allermeisten Fällen erreichen Kinder, die mit dem Edwards-Syndrom geboren werden, nicht das Erwachsenenalter. In der Vollform dieser Krankheit, wenn in jeder Körperzelle ein zusätzliches Chromosom vorhanden ist, sterben 90 % der Kinder vor dem Alter von einem Jahr aufgrund schwerwiegender Anomalien in der Entwicklung innerer Organe. Auch bei chirurgischer Korrektur möglicher Defekte und hochwertiger Pflege ist ihr Körper anfälliger für Infektionskrankheiten. Begünstigt wird dies auch durch Essstörungen, die bei den meisten Kindern auftreten. All dies erklärt die höchste Sterblichkeitsrate beim Edwards-Syndrom.
Bei einer milderen Mosaikform, bei der nur ein Teil der Zellen im Körper einen abnormalen Chromosomensatz enthält, ist die Überlebensrate etwas höher. Allerdings erreichen auch in diesen Fällen nur wenige Patienten das Erwachsenenalter. Ihre Aussehen bestimmt durch angeborene Anomalien, die bei der Geburt vorhanden waren ( Lippenspalte, deformiertes Ohr usw.). Das Hauptsymptom, das ausnahmslos bei allen Kindern auftritt, ist eine schwere geistige Behinderung. Wenn ein Kind mit Edwards-Syndrom das Erwachsenenalter erreicht hat, ist es zutiefst geistig zurückgeblieben ( IQ unter 20, was dem schwersten Grad geistiger Behinderung entspricht). Generell werden in der medizinischen Fachliteratur Einzelfälle beschrieben, bei denen Kinder mit Edwards-Syndrom das Erwachsenenalter erreichten. Aus diesem Grund liegen zu wenig objektive Daten vor, um über die äußeren Anzeichen dieser Krankheit bei Erwachsenen zu sprechen.
Diagnose genetischer Pathologie
 Derzeit gibt es drei Hauptstadien bei der Diagnose des Edwards-Syndroms, die jeweils mehrere mögliche Methoden umfassen. Da diese Krankheit unheilbar ist, sollten Eltern auf die Möglichkeiten dieser Methoden achten und diese nutzen. Die meisten Tests werden in speziellen Pränataldiagnostikzentren durchgeführt, in denen alle notwendigen Geräte zur Suche nach genetisch bedingten Erkrankungen vorhanden sind. Allerdings kann auch eine Beratung durch einen Genetiker oder Neonatologen sinnvoll sein.
Derzeit gibt es drei Hauptstadien bei der Diagnose des Edwards-Syndroms, die jeweils mehrere mögliche Methoden umfassen. Da diese Krankheit unheilbar ist, sollten Eltern auf die Möglichkeiten dieser Methoden achten und diese nutzen. Die meisten Tests werden in speziellen Pränataldiagnostikzentren durchgeführt, in denen alle notwendigen Geräte zur Suche nach genetisch bedingten Erkrankungen vorhanden sind. Allerdings kann auch eine Beratung durch einen Genetiker oder Neonatologen sinnvoll sein.
Die Diagnose des Edwards-Syndroms ist in folgenden Stadien möglich:
- Diagnose vor der Empfängnis;
- Diagnostik während der intrauterinen Entwicklung;
- Diagnose nach der Geburt.
Diagnose vor der Empfängnis
Die Diagnostik vor der Empfängnis eines Kindes ist eine ideale Option, aber leider sind ihre Möglichkeiten im gegenwärtigen Entwicklungsstadium der Medizin sehr begrenzt. Ärzte können mehrere Methoden anwenden, um auf eine erhöhte Wahrscheinlichkeit hinzuweisen, ein Kind mit einer Chromosomenstörung zu bekommen, aber nichts weiter. Tatsache ist, dass beim Edwards-Syndrom grundsätzlich keine Störungen bei den Eltern festgestellt werden können. Eine defekte Keimzelle mit 24 Chromosomen ist nur eine von vielen Tausenden. Deshalb kann man vor dem Zeitpunkt der Empfängnis nicht mit Sicherheit sagen, ob ein Kind mit dieser Krankheit zur Welt kommt.
Die wichtigsten diagnostischen Methoden vor der Empfängnis sind:
- Familiengeschichte. Eine Familienanamnese ist eine detaillierte Befragung beider Elternteile zu ihrer Abstammung. Der Arzt interessiert sich für alle Fälle erblicher ( und vor allem chromosomal) Krankheiten in der Familie. Wenn sich mindestens ein Elternteil an einen Fall von Trisomie erinnert ( Edwards-, Down-, Patau-Syndrom), erhöht dies die Wahrscheinlichkeit, ein krankes Kind zu bekommen, erheblich. Das Risiko beträgt jedoch immer noch nicht mehr als 1 %. Bei wiederholten Fällen dieser Erkrankungen bei Vorfahren steigt das Risiko um ein Vielfaches. Im Wesentlichen läuft die Analyse auf eine Konsultation mit einem Neonatologen oder Genetiker hinaus. Vorab können Eltern versuchen, detailliertere Informationen über ihre Vorfahren zu sammeln ( vorzugsweise 3 – 4 Knie). Dadurch wird die Genauigkeit dieser Methode erhöht.
- Erkennung von Risikofaktoren. Der Hauptrisikofaktor, der das Risiko für Chromosomenanomalien objektiv erhöht, ist das Alter der Mutter. Wie oben erwähnt, steigt für Mütter ab dem 40. Lebensjahr die Wahrscheinlichkeit, ein Kind mit Edwards-Syndrom zu bekommen, um ein Vielfaches. Berichten zufolge wurde nach 45 Jahren ( Alter der Mutter) geht fast jede fünfte Schwangerschaft mit einer Chromosomenpathologie einher. Die meisten davon enden mit einer Fehlgeburt. Weitere Faktoren sind frühere Infektionskrankheiten, chronische Krankheiten und schlechte Gewohnheiten. Ihre Rolle bei der Diagnose ist jedoch viel geringer. Auch auf die Frage, ob ein Kind mit Edwards-Syndrom gezeugt wird, gibt diese Methode keine genaue Antwort.
- Genetische Analyse der Eltern. Beschränkten sich frühere Methoden auf die Befragung der Eltern, handelt es sich bei der Genanalyse um eine vollwertige Studie, die spezielle Geräte, Reagenzien und qualifizierte Fachkräfte erfordert. Den Eltern wird Blut entnommen, aus dem im Labor Leukozyten isoliert werden. Nach der Behandlung mit speziellen Substanzen werden in diesen Zellen Chromosomen im Teilungsstadium deutlich sichtbar. Auf diese Weise wird der Karyotyp der Eltern ermittelt. In den allermeisten Fällen ist es normal ( Bei den hier nachweisbaren Chromosomenanomalien ist die Wahrscheinlichkeit einer Fortpflanzung vernachlässigbar). Darüber hinaus verwenden Sie spezielle Marker ( Fragmente von Molekülketten) DNA-Abschnitte mit defekten Genen können erkannt werden. Hierbei handelt es sich jedoch nicht um Chromosomenanomalien, sondern um genetische Mutationen, die keinen direkten Einfluss auf die Wahrscheinlichkeit eines Edwards-Syndroms haben. Daher liefert die genetische Analyse der Eltern vor der Empfängnis trotz der Komplexität und der hohen Kosten auch keine eindeutige Antwort auf die Prognose dieser Pathologie.
Diagnose während der fetalen Entwicklung
Während der intrauterinen Entwicklung gibt es mehrere Methoden, die das Vorliegen einer Chromosomenpathologie beim Fötus direkt oder indirekt bestätigen können. Die Genauigkeit dieser Methoden ist viel höher, da sich Ärzte nicht mit den Eltern, sondern mit dem Fötus selbst befassen. Zur Untersuchung stehen sowohl der Embryo selbst als auch seine Zellen mit eigener DNA zur Verfügung. Dieses Stadium wird auch Pränataldiagnostik genannt und ist das wichtigste. Zu diesem Zeitpunkt können Sie die Diagnose bestätigen, die Eltern vor dem Vorliegen einer Pathologie warnen und gegebenenfalls die Schwangerschaft abbrechen. Wenn sich eine Frau für die Geburt entscheidet und das Neugeborene noch am Leben ist, haben die Ärzte die Möglichkeit, sich im Voraus darauf vorzubereiten, ihm die notwendige Pflege zukommen zu lassen.
Die wichtigsten Forschungsmethoden im Rahmen der Pränataldiagnostik sind:
- Ultraschall ( Ultraschall) . Diese Methode ist nicht-invasiv, das heißt, das Gewebe der Mutter oder des Fötus wird nicht geschädigt. Es ist völlig sicher und wird im Rahmen der Pränataldiagnostik allen schwangeren Frauen empfohlen ( unabhängig von ihrem Alter oder einem erhöhten Risiko für Chromosomenerkrankungen). Das Standardprogramm geht davon aus, dass der Ultraschall dreimal durchgeführt werden sollte ( in der 10. – 14., 20. – 24. und 32. – 34. Schwangerschaftswoche). Wenn der behandelnde Arzt die Möglichkeit angeborener Fehlbildungen vermutet, können außerplanmäßige Ultraschalluntersuchungen durchgeführt werden. Das Edwards-Syndrom kann durch eine Verzögerung der Größe und des Gewichts des Fötus, eine große Menge Fruchtwasser und sichtbare Entwicklungsanomalien angezeigt werden ( Mikrozephalie, Knochenverformung). Diese Störungen weisen mit hoher Wahrscheinlichkeit auf schwere genetische Erkrankungen hin, das Edwards-Syndrom kann jedoch nicht definitiv bestätigt werden.
- Amniozentese. Amniozentese ist eine zytologische ( zellular) Analyse von Fruchtwasser. Der Arzt führt unter der Kontrolle eines Ultraschallgeräts vorsichtig eine spezielle Nadel ein. Die Punktion erfolgt an einer Stelle, an der sich keine Nabelschnurschlingen befinden. Mit einer Spritze wird die für die Untersuchung benötigte Menge Fruchtwasser entnommen. Der Eingriff kann in allen Schwangerschaftstrimestern durchgeführt werden, der optimale Zeitraum zur Diagnose von Chromosomenstörungen ist jedoch der Zeitraum nach der 15. Schwangerschaftswoche. Komplikationsrate ( bis hin zur spontanen Abtreibung) beträgt bis zu 1 %, daher sollte der Eingriff bei fehlender Indikation nicht durchgeführt werden. Nach dem Sammeln von Fruchtwasser wird das resultierende Material verarbeitet. Diese Flüssigkeiten enthalten Zellen von der Hautoberfläche des Babys, die Proben seiner DNA enthalten. Sie sind diejenigen, die auf das Vorliegen genetischer Krankheiten getestet werden.
- Cordozentese. Die Cordozentese ist die aussagekräftigste Methode der Pränataldiagnostik. Nach der Narkose und unter Kontrolle eines Ultraschallgeräts durchsticht der Arzt mit einer speziellen Nadel das durch die Nabelschnur verlaufende Gefäß. So wird eine Blutprobe entnommen ( bis zu 5 ml) sich entwickelndes Kind. Die Technik zur Durchführung der Analyse ähnelt der für Erwachsene. Dieses Material kann mit hoher Genauigkeit auf verschiedene genetische Anomalien untersucht werden. Dazu gehört auch die fetale Karyotypisierung. Liegt ein zusätzliches 18. Chromosom vor, kann man von einem gesicherten Edwards-Syndrom sprechen. Dieser Test wird nach der 18. Schwangerschaftswoche empfohlen ( optimal 22 – 25 Wochen). Die Häufigkeit möglicher Komplikationen nach einer Cordozentese beträgt 1,5 – 2 %.
- Chorionzottenbiopsie. Das Chorion ist eine der embryonalen Membranen, die Zellen mit der genetischen Information des Fötus enthält. Bei dieser Studie wird die Gebärmutter unter Narkose durch die vordere Bauchdecke punktiert. Mit einer speziellen Biopsiezange wird eine Gewebeprobe zur Analyse entnommen. Anschließend wird eine standardmäßige genetische Untersuchung des gewonnenen Materials durchgeführt. Zur Diagnose des Edwards-Syndroms wird eine Karyotypisierung durchgeführt. Als optimaler Zeitraum für die Durchführung einer Chorionzottenbiopsie gilt die 9.–12. Schwangerschaftswoche. Die Komplikationsrate beträgt 2 – 3 %. Der Hauptvorteil, der es von anderen Methoden unterscheidet, ist die Geschwindigkeit, mit der Ergebnisse erzielt werden ( innerhalb von 2 – 4 Tagen).
Diagnose nach der Geburt
Die Diagnose des Edwards-Syndroms nach der Geburt ist am einfachsten, schnellsten und genauesten. Leider wurde zu diesem Zeitpunkt bereits ein Kind mit einer schweren genetischen Pathologie geboren, wirksame Behandlung was es in unserer Zeit noch nicht gibt. Wenn die Krankheit im Stadium der pränatalen Diagnose nicht erkannt wurde ( oder entsprechende Studien wurden nicht durchgeführt), dann besteht unmittelbar nach der Geburt der Verdacht auf ein Edwards-Syndrom. Das Baby ist in der Regel ausgetragen oder sogar nach der Geburt geboren, aber sein Gewicht liegt immer noch unter dem Durchschnitt. Darüber hinaus sind einige der oben genannten Geburtsfehler bemerkenswert. Wenn sie bemerkt werden, wird ein Gentest durchgeführt, um die Diagnose zu bestätigen. Dem Kind wird Blut zur Analyse entnommen. Allerdings ist die Bestätigung des Vorliegens eines Edwards-Syndroms in diesem Stadium nicht das Hauptproblem.
Die Hauptaufgabe bei der Geburt eines Kindes mit dieser Pathologie besteht darin, Anomalien in der Entwicklung der inneren Organe zu erkennen, die in der Regel in den ersten Lebensmonaten zum Tod führen. Die meisten diagnostischen Verfahren unmittelbar nach der Geburt zielen darauf ab, danach zu suchen.
Um Defekte in der Entwicklung innerer Organe zu erkennen, werden folgende Forschungsmethoden eingesetzt:
- Ultraschalluntersuchung der Bauchhöhle;
- Echokardiographie;
- allgemeiner Bluttest und biochemischer Bluttest;
- allgemeine Urinanalyse;
- Computertomographie oder Magnetresonanztomographie;
- Radiographie.
Für eine Röntgenuntersuchung gilt in der Regel das Säuglingsalter als Kontraindikation, in diesem Fall kann die Gefahr jedoch vernachlässigt werden. Tatsache ist, dass es im Moment darum geht, lebensgefährliche Pathologien sofort zu erkennen. Daher müssen Ärzte dringend alle notwendigen Informationen über bestehende Fehlbildungen einholen. Dies wird Ihnen bei der Auswahl der Behandlungstaktiken helfen. In den meisten Fällen tragen eine kompetente Patientenbetreuung und eine angemessene Pflege dazu bei, das Leben des Kindes zu verlängern.
Daraus können wir schließen, dass es viele Methoden zur Diagnose des Edwards-Syndroms gibt. Einige von ihnen ( Amniozentese, Cordozentese usw.) bergen ein gewisses Komplikationsrisiko und werden nicht ohne besondere Indikation durchgeführt. Die Hauptindikationen sind das Vorliegen von Fällen von Chromosomenerkrankungen in der Familie und das Alter der Mutter über 35 Jahre. Das Diagnose- und Behandlungsprogramm für die Patientin in allen Stadien der Schwangerschaft kann vom behandelnden Arzt bei Bedarf geändert werden.
Prognose für Kinder mit Edwards-Syndrom
 Aufgrund der vielfältigen Entwicklungsstörungen, die das Edwards-Syndrom mit sich bringt, ist die Prognose für Neugeborene mit dieser Diagnose fast immer ungünstig. Statistische Daten ( aus verschiedenen unabhängigen Studien) Sie sagen, dass mehr als die Hälfte der Kinder ( 50 – 55%
) werden nicht älter als drei Monate. Weniger als zehn Prozent der Babys schaffen es, ihren ersten Geburtstag zu feiern. Kinder, die ein höheres Lebensalter erreichen, haben schwerwiegende gesundheitliche Probleme und benötigen ständige Pflege. Um das Leben zu verlängern, sind oft aufwendige chirurgische Eingriffe am Herzen, an den Nieren oder anderen inneren Organen notwendig. Die Korrektur von Geburtsfehlern und die fortlaufende fachmännische Betreuung sind im Wesentlichen die einzige Behandlung. Bei Kindern mit der klassischen Form des Edwards-Syndroms ( komplette Trisomie 18) gibt es praktisch keine Chance auf eine normale Kindheit oder ein langes Leben.
Aufgrund der vielfältigen Entwicklungsstörungen, die das Edwards-Syndrom mit sich bringt, ist die Prognose für Neugeborene mit dieser Diagnose fast immer ungünstig. Statistische Daten ( aus verschiedenen unabhängigen Studien) Sie sagen, dass mehr als die Hälfte der Kinder ( 50 – 55%
) werden nicht älter als drei Monate. Weniger als zehn Prozent der Babys schaffen es, ihren ersten Geburtstag zu feiern. Kinder, die ein höheres Lebensalter erreichen, haben schwerwiegende gesundheitliche Probleme und benötigen ständige Pflege. Um das Leben zu verlängern, sind oft aufwendige chirurgische Eingriffe am Herzen, an den Nieren oder anderen inneren Organen notwendig. Die Korrektur von Geburtsfehlern und die fortlaufende fachmännische Betreuung sind im Wesentlichen die einzige Behandlung. Bei Kindern mit der klassischen Form des Edwards-Syndroms ( komplette Trisomie 18) gibt es praktisch keine Chance auf eine normale Kindheit oder ein langes Leben.
Bei partieller Trisomie oder Mosaikform des Syndroms ist die Prognose etwas besser. Die durchschnittliche Lebenserwartung steigt auf mehrere Jahre. Dies erklärt sich dadurch, dass Entwicklungsanomalien bei milderen Formen nicht so schnell zum Tod des Kindes führen. Das Hauptproblem, nämlich die schwere geistige Behinderung, ist jedoch ausnahmslos allen Patienten gemeinsam. Mit Erreichen der Pubertät besteht keine Chance mehr auf Fortpflanzung ( Die Pubertät findet in der Regel nicht statt), noch auf die Möglichkeit zu arbeiten ( sogar mechanisch, was keine besonderen Fähigkeiten erfordert). Für die Betreuung von Kindern mit angeborenen Erkrankungen gibt es spezielle Zentren, in denen Patienten mit Edwards-Syndrom betreut und wenn möglich unterstützt werden intellektuelle Entwicklung. Bei ausreichender Anstrengung von Ärzten und Eltern kann ein Kind, das länger als ein Jahr lebt, lernen, zu lächeln, auf Bewegungen zu reagieren, die Körperhaltung selbstständig beizubehalten oder zu essen ( in Abwesenheit von Defekten des Verdauungssystems). Daher sind weiterhin Anzeichen einer Entwicklung zu beobachten.
Die hohe Kindersterblichkeitsrate bei dieser Krankheit erklärt sich aus einer Vielzahl von Fehlbildungen innerer Organe. Sie sind bei der Geburt sofort unsichtbar, sind aber bei fast allen Patienten vorhanden. In den ersten Lebensmonaten sterben Kinder meist an Herz- oder Atemstillstand.
Am häufigsten werden Entwicklungsstörungen in folgenden Organen und Systemen beobachtet:
- Bewegungsapparat ( Knochen und Gelenke, einschließlich des Schädels);
- das Herz-Kreislauf-System;
- zentrales Nervensystem;
- Verdauungssystem;
- Urogenitalsystem;
- andere Verstöße.
Bewegungsapparat
Die Hauptdefekte in der Entwicklung des Bewegungsapparates sind Fehlstellungen der Finger und Krümmungen der Füße. Am Hüftgelenk werden die Beine so zusammengeführt, dass sich die Knie fast berühren und die Füße leicht zur Seite schauen. Es ist nicht ungewöhnlich, dass Kinder mit Edwards-Syndrom ein ungewöhnlich kurzes Brustbein haben. Dies führt zu einer Verformung des gesamten Brustkorbs und zu Atemproblemen, die sich mit zunehmendem Alter verschlimmern, auch wenn die Lunge selbst nicht betroffen ist.
Defekte in der Entwicklung des Schädels sind hauptsächlich kosmetischer Natur. Defekte wie Gaumenspalten, Lippenspalten und Gaumenspalten führen jedoch zu ernsthaften Schwierigkeiten bei der Ernährung des Kindes. Oft wird das Kind vor Operationen zur Korrektur dieser Defekte auf parenterale Ernährung umgestellt ( in Form von Tropfern mit Nährlösungen). Eine weitere Möglichkeit ist die Verwendung einer Gastrostomiesonde, einem speziellen Schlauch, durch den die Nahrung direkt in den Magen gelangt. Seine Installation erfordert einen separaten chirurgischen Eingriff.
Im Allgemeinen stellen Fehlbildungen des Bewegungsapparates keine unmittelbare Gefahr für das Leben des Kindes dar. Sie wirken sich jedoch indirekt auf sein Wachstum und seine Entwicklung aus. Die Häufigkeit solcher Veränderungen liegt bei Patienten mit Edwards-Syndrom bei etwa 98 %.
Das Herz-Kreislauf-System
Fehlbildungen des Herz-Kreislauf-Systems sind die häufigste Todesursache im frühen Kindesalter Kindheit. Tatsache ist, dass solche Verstöße in fast 90 % der Fälle vorkommen. Meistens stören sie den Bluttransport durch den Körper ernsthaft und führen zu schwerer Herzinsuffizienz. Die meisten Herzerkrankungen können chirurgisch korrigiert werden, aber nicht jedes Kind kann sich einer so komplexen Operation unterziehen.
Die häufigsten Anomalien des Herz-Kreislauf-Systems sind:
- Nichtverschluss des Vorhofseptums;
- Nichtverschluss des interventrikulären Septums;
- Verschmelzung der Klappensegel ( oder umgekehrt ihre Unterentwicklung);
- Coarktation ( Verengung) Aorta.
Alle diese Herzfehler führen zu schwerwiegenden Durchblutungsstörungen. Arterielles Blut fließt nicht in der erforderlichen Menge in das Gewebe, weshalb die Körperzellen abzusterben beginnen.
zentrales Nervensystem
Verdauungssystem
Häufigkeit von Mängeln Verdauungssystem beim Edwards-Syndrom sind es bis zu 55 %. In den meisten Fällen stellen diese Entwicklungsanomalien eine ernsthafte Bedrohung für das Leben des Kindes dar, da sie es ihm nicht ermöglichen, Nährstoffe richtig aufzunehmen. Das Essen unter Umgehung der natürlichen Verdauungsorgane schwächt den Körper erheblich und verschlimmert den Zustand des Kindes.
Die häufigsten Fehlbildungen des Verdauungssystems sind:
- Meckel-Divertikel ( Blinddarm im Dünndarm);
- Ösophagusatresie ( Überwucherung seines Lumens, wodurch die Nahrung nicht in den Magen gelangt);
- Gallenatresie ( Ansammlung von Galle in der Blase).
Alle diese Pathologien erfordern eine chirurgische Korrektur. In den meisten Fällen hilft eine Operation nur geringfügig, das Leben des Kindes zu verlängern.
Urogenitalsystem
Andere Verstöße
Weitere mögliche Entwicklungsstörungen sind Hernien ( Nabelschnur, Leistengegend). Es können auch Bandscheibenvorfälle der Wirbelsäule festgestellt werden, die zu neurologischen Problemen führen können. Manchmal wird Mikrophthalmie in den Augen beobachtet ( kleine Augapfelgröße).
Die Kombination dieser Entwicklungsstörungen führt zu einer hohen Kindersterblichkeit. Wenn das Edwards-Syndrom früh in der Schwangerschaft diagnostiziert wird, raten Ärzte in den meisten Fällen aus medizinischen Gründen zu einem Schwangerschaftsabbruch. Die endgültige Entscheidung trifft jedoch die Patientin selbst. Trotz der Schwere der Erkrankung und der schlechten Prognose hoffen viele lieber auf das Beste. Leider sind in naher Zukunft keine größeren Änderungen bei den Methoden zur Diagnose und Behandlung des Edwards-Syndroms zu erwarten.
- Wesen der Krankheit
- Ursachen
- Symptome
- Diagnose
- Behandlung
- Prognosen
Nach dem Down-Syndrom ist das Edwards-Syndrom die häufigste Chromosomenstörung. Da die Prognose für Kinder, die an dieser Pathologie leiden, am enttäuschendsten ist (Tod oder tiefe geistige Behinderung für den Rest ihres Lebens), müssen Eltern unbedingt wissen, um welche Art von Abweichung es sich handelt und wann sie diagnostiziert werden kann, um die Diagnose stellen zu können rechtzeitig die richtige Entscheidung zu treffen, ob ein solches Baby im Mutterleib belassen oder die Schwangerschaft abgebrochen werden soll.
Wesen der Krankheit
Das Trisomie-18-Syndrom oder Edwards-Syndrom ist eine Chromosomenerkrankung, die durch einen ganzen Komplex multipler, sehr schwerwiegender Entwicklungsstörungen gekennzeichnet ist. Der Grund ist die Bildung eines zusätzlichen 18. Chromosoms. Somit besteht der Karyotyp eines Patienten mit Edwards-Syndrom aus drei 18. Chromosomen statt der normalen zwei. In 90 % der Fälle handelt es sich um eine einfache Trisomie, seltener um ein Mosaik oder eine Translokation.
Die Formel, nach der der Karyotyp des Edwards-Syndroms gelesen wird, lautet wie folgt: 47,XX, 18+ (das ist ein Mädchen) und 47,XY, 18+ (das ist ein Junge).
Durch die Seiten der Geschichte. Das Edwards-Syndrom wurde erstmals 1960 von John Edwards beschrieben.
Ursachen
Die Hauptursache der Erkrankung ist das verdreifachte (und nicht wie üblich verdoppelte) Chromosom 18 im Karyotyp der Zygote. Was genau diese Pathologie hervorruft, ist den Genetikern jedoch noch unbekannt. Darüber hinaus entsteht bereits vor der Befruchtung ein zusätzliches Chromosom. Unter Ärzten herrscht die Meinung vor, dass keine anderen Tatsachen als der bloße Zufall dafür verantwortlich sind. Andererseits gibt es Risikofaktoren, die eine Eingrenzung der angeblichen Ursachen des Edwards-Syndroms in eine besondere Gruppe ermöglichen, die Paare mit Kinderwunsch im Auge behalten müssen.
- Das Alter der Mutter beträgt mehr als 45 Jahre. In diesem Fall beträgt das Risiko eines Edwards-Syndroms 0,7 %.
- Vererbung: Wenn in der Familie bereits Kinder mit ähnlichen Anomalien geboren wurden, besteht ein sehr hohes Risiko, ein Kind mit einer Pathologie des Karyotyps zur Welt zu bringen.
- Auch hier bleiben schlechte Gewohnheiten bestehen. Einige Wissenschaftler beweisen, dass die Mosaikform des Edwards-Syndroms (es ist die mildeste) durch langfristigen Drogenkonsum, eine große Menge Nikotin oder Alkohol im Körper eines Elternteils hervorgerufen werden kann.
- Langfristige Einnahme wirksamer Medikamente.
- Infektionen des Genitaltrakts.
- Strahlenbelastung führt sehr häufig zu Chromosomenmutationen.
Dies sind nur die von Genetikern vorgeschlagenen Gründe für das Auftreten von drei statt zwei 18. Chromosomen. Sie sollten nicht als unerschütterliches Postulat verstanden werden. Risikofaktoren für das Edwards-Syndrom sind demnach das Alter der Mutter, bereits in der Familie bestehende Chromosomenerkrankungen und schlechte Angewohnheiten.
Im Vergleich zum Down-Syndrom kommt diese Pathologie eher selten vor. Viele Eltern fragen sich, wie oft das Edwards-Syndrom diagnostiziert wird: Laut Statistik wird von 7.000 gesunden Babys 1 krankes Kind geboren. Da die Prognose für solche Kinder völlig enttäuschend ist, ist es besser, sich vorab über die Diagnose zu informieren.
Interessante Tatsache. Es ist immer noch unerklärlich, aber es werden dreimal mehr Mädchen mit dem Edwards-Syndrom geboren als Jungen.
Symptome
Um während der Schwangerschaft die richtige Entscheidung zu treffen, ob sie ihr Kind mit dieser Krankheit belassen oder nicht, müssen Eltern genau verstehen, wie Kinder mit Edwards-Syndrom bei der Geburt sind. Krankheitsbild Die Pathologie ist düster und schwer für das Herz der Mutter und des Vaters. Die Hauptsymptome sind bereits ab der Geburt des Babys erkennbar.
Bei der Geburt:
- geringes Gewicht (ca. 2 kg) und vorgeburtliche Unterernährung während der normalen und sogar längeren Schwangerschaftsdauer;
- Erstickung.
Verformungen von Gesicht und Schädel:
- Anomalien des Schädels, der eine dolichozephale Form, eine niedrige Stirn und einen hervorstehenden Hinterkopf hat;
- sehr kleine Mundöffnung und Unterkiefer;
- Gaumen- und Oberlippenspalte;
- Strabismus;
- Verformung der Ohren, ihre niedrige Lage relativ zum Gesicht, Dehnung in der horizontalen Ebene;
- Fehlen von Lappen und Tragus;
- verengter äußerer Gehörgang, in manchen Fällen fehlt er vollständig;
- Ptosis – schlaffe Haut;
- kurzer Hals mit charakteristischer Kragenhautfalte.
Skelettdeformitäten:
- kurzes Brustbein, wodurch die Interkostalräume kleiner werden und die Brust kürzer und breiter als normal ist;
- in 80% der Fälle - abnormale Entwicklung Füße: Die Fersen ragen stark hervor, das Fußgewölbe hängt durch (Schaukelfüße), die großen Zehen sind verdickt und verkürzt;
- gekreuzte Finger;
- angeborene Hüftluxation;
- Klumpfuß.
Pathologien innerer Organe:
- Herz- und Gefäßfehler;
- Kleinhirnhypoplasie;
- Magen-Darm-Erkrankungen;
- Probleme mit dem Urogenitalsystem;
- Funktionsstörungen des Zentralnervensystems;
- ausgeprägte geistige Behinderung.
Allgemeine Entwicklung:
- Schwierigkeiten beim Schlucken, Saugen und Atmen.
Ein Kind mit Edwards-Syndrom wird nie ein erfülltes Leben wie alle anderen führen können. In seinem kurzen Leben wird er viele Hindernisse überwinden und sich seiner Krankheit überhaupt nicht bewusst sein. Das Schwierigste ist für Eltern, die täglich die Entwicklung von Pathologien und Defekten beobachten müssen, mit denen ihr Baby geboren wurde. Wann wird diese Krankheit diagnostiziert?
Wow! Der Genetiker Edwards, der diese Krankheit beschrieb, identifizierte über 130 symptomatische Defekte, die für dieses Syndrom charakteristisch sind.
Diagnose
Die Diagnose des Edwards-Syndroms ist sehr wichtig, da diese Chromosomenpathologie eine medizinische Indikation für einen Schwangerschaftsabbruch darstellt. Die folgenden Methoden werden zur Identifizierung der Krankheit verwendet.
Ultraschall und Dopplerographie
Es gibt bestimmte Anzeichen des Edwards-Syndroms im Ultraschall, die Ihr Arzt beachten sollte:
- multiple fetale Entwicklungsanomalien;
- Agenesie der Nabelarterie;
- kleine Plazenta;
- Polyhydramnion.
All dies sind jedoch indirekte Anzeichen des Edwards-Syndroms während der Schwangerschaft, die durch Daten aus anderen Studien bestätigt werden müssen.
Standardmäßiges pränatales Screening
Beinhaltet eine Blutuntersuchung auf das Vorhandensein von Serummarkern:
- von 11 bis 13 Wochen: βhCG und PAPP;
- 20 bis 24 Wochen: βhCG, freies Östriol, α-Fetoprotein.
Fällt eine Schwangere aufgrund dieser Tests in eine Hochrisikogruppe, wird ihr eine zusätzliche Diagnostik angeboten.
Invasive Diagnosemethoden
Sie werden in Ausnahmefällen durchgeführt, wenn das Risiko, ein Kind mit Edwards-Syndrom zu bekommen, sehr hoch ist. Zur invasiven Diagnostik gehören:
- Chorionzottenbiopsie;
- Amniozentese;
- Cordozentese;
- anschließende fetale Karyotypisierung.
Wenn die Krankheit nicht rechtzeitig erkannt wurde und ein Kind mit Edwards-Syndrom noch geboren wird, wird es sofort einer umfassenden Untersuchung unterzogen.
Diagnose nach der Geburt
Die Untersuchung eines kranken Kindes zielt darauf ab, Entwicklungsstörungen festzustellen. Ein Neugeborenes mit Edwards-Syndrom wird von verschiedenen Spezialisten untersucht:
- Neonatologe;
- Kardiologe;
- Neurologe;
- der Chirurg;
- Orthopäde;
- Urologe usw.
Die wichtigsten diagnostischen Tests, die bei einem Kind mit Edwards-Syndrom unmittelbar nach der Geburt durchgeführt werden, sind:
- Echokardiographie;
- Ultraschall der Nieren;
- Ultraschall der Bauchorgane.
Der Schwangerschaftsverlauf bei der Geburt eines Kindes mit Edwards-Syndrom unterscheidet sich in keiner Weise. Er beginnt sich auch rechtzeitig zu bewegen und ist sogar sehr aktiv; eine zusätzliche Toxikose ist nicht zu beobachten. Daher können in diesem Zeitraum nur moderne Diagnosegeräte die Krankheit erkennen. Wenn das Baby noch geboren wird, wird sein gesamtes kurzes Leben mit der ständigen Behandlung der damit verbundenen Defekte verbracht.
Meinung der Ärzte. Es werden immer mehr Mädchen mit dem Edwards-Syndrom geboren, nicht weil sie am häufigsten für diese Pathologie auf Chromosom 18 anfällig sind. Der Grund ist ihr Überleben. Es wird angenommen, dass die meisten Schwangerschaften bei Jungen mit diesem Syndrom mit einem Spontanabort oder dem intrauterinen Tod des Fötus enden.
Behandlung
Bei solchen Kindern sind Entwicklungsanomalien und alle Arten von Defekten nicht mit dem Leben vereinbar, sodass die Behandlung des Edwards-Syndroms auf eine symptomatische Behandlung beschränkt ist. Ziel ist es, grundlegende physiologische Funktionen aufrechtzuerhalten, die Lebensqualität zu verbessern und so weit wie möglich zu verlängern. Die chirurgische Korrektur angeborener Pathologien ist nach Ansicht der meisten Ärzte ungerechtfertigt und sehr riskant. In den ersten Lebenstagen beschränkt sich die Behandlung auf folgende Aktivitäten:
- Sondenernährung aufgrund von Schluck- und Saugbeschwerden;
- langfristige mechanische Beatmung aufgrund von Atemproblemen.
Nach der Entlassung sollten Eltern dem kranken Baby (wenn möglich) Folgendes zur Verfügung stellen:
- organisierte, fast professionelle Betreuung: Es ist gut, wenn neben dem Kind eine Person (Nanny) mit medizinischer Ausbildung steht;
- gute Ernährung;
- Regelmäßige Überwachung durch verschiedene Kinderärzte.
Wir müssen sofort bedenken, dass die Betreuung eines Kindes mit Edwards-Syndrom viel Geduld und Kraft erfordert.
Prognosen
Eltern, deren Kind Träger des Edwards-Syndroms ist, sollten wissen, welche Prognose sie erwartet. Wie oben erwähnt sind sie sehr enttäuschend:
- die Lebenserwartung solcher Kinder ist sehr kurz;
- 60 % sterben vor dem dritten Lebensmonat (Jungen fallen am häufigsten in diese Gruppe);
- weitere 10 % überleben bis zu einem Jahr;
- Nur 1 % der Geborenen erreichen das 10. Lebensjahr;
- die häufigste Todesursache ist eine Herzfunktionsstörung oder ein Atemstillstand;
- alle Kinder mit Edwards-Syndrom bleiben bis zum Ende ihrer Tage stark oligofren;
- der Körper solcher Kinder ist sehr geschwächt und anfällig für alle möglichen Krankheiten, weshalb sie häufig an Harnwegsinfektionen, Bindehautentzündung, Mittelohrentzündung, Sinusitis und Lungenentzündung leiden;
- Die verbalen Kommunikationsfähigkeiten solcher Kinder sind sehr begrenzt;
- sie können auf die Worte ihrer Eltern reagieren;
- manche lächeln sogar, erkennen Betreuer und interagieren mit ihnen;
- in der Lage, Fähigkeiten wie Selbsternährung und Kopfheben zu erwerben.
Wenn bei einem Kind während der Schwangerschaft das Edwards-Syndrom diagnostiziert wurde, zwingt dies die Eltern zu einer verantwortungsvollen Entscheidung (den Zeitpunkt der Diagnose verschiedener fetaler Pathologien finden Sie im nächsten Artikel). Werden sie in der Lage sein, ein schwerkrankes Kind zu betreuen, das an mehreren Pathologien leidet? Oder ist es sinnvoll, eine solche Schwangerschaft abzubrechen? Hier kann nur das Paar selbst durch gemeinsame Anstrengungen den einzig richtigen Ausweg für sich finden.
Während der Schwangerschaft unterziehen sich werdende Mütter einem pränatalen Screening, um genetische Anomalien beim Fötus frühzeitig zu erkennen. Eine der schwersten Erkrankungen dieser Gruppe ist das 1960 von John Edwards beschriebene Syndrom. Medizinisch spricht man von Trisomie 18.
Edwards-Syndrom – was ist das in einfachen Worten?
Eine gesunde männliche und weibliche Fortpflanzungszelle verfügt über einen Standard- oder haploiden Chromosomensatz in Höhe von 23 Stück. Nach der Verschmelzung bilden sie einen individuellen Satz – einen Karyotyp. Es handelt sich um eine Art DNA-Pass, der einzigartige genetische Daten über das Kind enthält. Ein normaler oder diploider Karyotyp enthält 46 Chromosomen, 2 von jedem Typ, von der Mutter und dem Vater.
Bei der betrachteten Krankheit gibt es im 18. Paar ein zusätzliches dupliziertes Element. Dies ist Trisomie oder Edwards-Syndrom – ein Karyotyp, der aus 47 statt 46 Chromosomen besteht. Manchmal ist die dritte Kopie von Chromosom 18 teilweise oder nicht in allen Zellen vorhanden. Solche Fälle werden selten diagnostiziert (etwa 5 %), diese Nuancen haben keinen Einfluss auf den Verlauf der Pathologie.
Edwards-Syndrom – Ursachen
Die Genetik hat noch nicht herausgefunden, warum die beschriebene Chromosomenmutation bei einigen Kindern auftritt. Es wird davon ausgegangen, dass es sich um einen Unfall handelt, und es wurden keine vorbeugenden Maßnahmen entwickelt, um dies zu verhindern. Einige Experten assoziieren externe Faktoren mit dem Edwards-Syndrom – Gründe, die vermutlich zur Entstehung der Anomalie beitragen:
- langfristiger Konsum von Drogen, Alkohol, Tabak;
- Vererbung;
- das Alter der Mutter oder des Vaters beträgt über 45 Jahre;
- Genitalinfektionen;
- Langzeiteinnahme von Medikamenten, die das Immunsystem, das Hormonsystem und das Hormonsystem beeinflussen Fortpflanzungsapparat;
- Exposition gegenüber radioaktiver Strahlung.
Edwards-Syndrom – Genetik
Aktuellen Studien zufolge enthält Chromosom 18 557 DNA-Abschnitte. Sie kodieren mehr als 289 Arten von Proteinen im Körper. IN Prozentsatz das sind 2,5-2,6 % des genetischen Materials, weshalb das dritte 18. Chromosom einen so starken Einfluss auf die Entwicklung des Fötus hat – das Edwards-Syndrom schädigt die Schädelknochen, das Herz-Kreislauf- und Urogenitalsystem. Die Mutation betrifft einige Teile des Gehirns und periphere Nervengeflechte. Ein Patient mit Edwards-Syndrom ist durch den in der Abbildung dargestellten Karyotyp gekennzeichnet. Es zeigt deutlich, dass alle Sätze gepaart sind, mit Ausnahme von Satz 18.
Inzidenz des Edwards-Syndroms
Diese Pathologie ist selten, insbesondere im Vergleich zu bekannteren genetischen Anomalien. Bei einem von 7.000 gesunden Babys, meist Mädchen, wird das Edwards-Syndrom diagnostiziert. Man kann nicht sagen, dass das Alter des Vaters oder der Mutter einen wesentlichen Einfluss auf die Wahrscheinlichkeit einer Trisomie 18 hat. Das Edwards-Syndrom tritt bei Kindern nur um 0,7 % häufiger auf, wenn die Eltern über 45 Jahre alt sind. Diese Chromosomenmutation kommt auch bei Babys vor, die in jungen Jahren gezeugt wurden.
Edwards-Syndrom - Anzeichen
Die betreffende Krankheit weist ein spezifisches Krankheitsbild auf, das eine genaue Identifizierung der Trisomie 18 ermöglicht. Es gibt zwei Gruppen von Anzeichen, die das Edwards-Syndrom begleiten – die Symptome werden herkömmlicherweise in innere Funktionsstörungen von Organen und äußere Anomalien eingeteilt. Die erste Art von Manifestationen umfasst:
- Nabel- und Leistenhernien;
- angeborene Herzfehler;
- Mangel an Saug- und Schluckreflex;
- Meckel-Divertikel;
- gastroösophagealer Reflux;
- Atresie des Anus oder der Speiseröhre;
- Klitorishypertrophie;
- Unterentwicklung des Corpus callosum, Kleinhirn;
- Kryptorchismus;
- falsche Lage des Darms;
- Hypospadie;
- Verdoppelung der Harnleiter;
- Atrophie oder Glättung der Gehirnwindungen;
- segmentierte oder hufeisenförmige Niere;
- ungeformte Eierstöcke;
- gebogener Rücken;
- Muskeldystrophie;
- geringes Körpergewicht (ca. 2 kg bei der Geburt).
Auch äußerlich ist das Edwards-Syndrom leicht zu erkennen – ein Foto von Babys mit Trisomie 18 zeigt das Vorhandensein folgender Anzeichen:
- unverhältnismäßig kleiner Kopf;
- verzerrte Gesichtsform;
- schmale und kurze Lidspalten;
- deformierte, tief liegende Ohren (horizontal gestreckt);
- Fehlen des Ohrläppchens, manchmal des Tragus und sogar des Gehörgangs;
- kurze und breite Brust;
- unterentwickelter Unterkiefer;
- kleiner Mund, hat aufgrund einer pathologisch verkürzten Oberlippe oft eine dreieckige Öffnung;
- deprimierter, verbreiterter Nasenrücken;
- „Wippfuß“;
- Membranen zwischen den Fingern oder deren Verschmelzung (flossenartige Gliedmaßen);
- hoher Gaumen, manchmal mit einer Spalte;
- kurzer Hals mit markanter Kragenfalte;
- Querrillen und Grate auf den Handflächen;
- Hämangiome und Papillome auf der Haut;
- Augenlid-Ptosis;
- Strabismus;
- hervorstehender Nacken und niedrige Stirn.
Edwards-Syndrom - Diagnose
Die beschriebene genetische Erkrankung stellt eine direkte Indikation für einen Schwangerschaftsabbruch dar. Kinder mit Edwards-Syndrom werden nie ein erfülltes Leben führen können und ihr Gesundheitszustand wird sich rapide verschlechtern. Aus diesem Grund ist es wichtig, die Diagnose Trisomie 18 so früh wie möglich zu stellen. früh. Zur Feststellung dieser Pathologie wurden mehrere informative Screenings entwickelt.
Test auf Edwards-Syndrom
Es gibt nicht-invasive und invasive Methoden zur Untersuchung biologischen Materials. Die zweite Art von Tests gilt als die zuverlässigste und zuverlässigste; sie hilft, das Edwards-Syndrom beim Fötus in den frühen Entwicklungsstadien zu erkennen. Das standardmäßige pränatale mütterliche Blutscreening ist nicht-invasiv. Zu den invasiven Diagnosemethoden gehören:
- Chorionzottenbiopsie. Die Studie wird ab 8 Wochen durchgeführt. Zur Durchführung der Analyse wird ein Stück der Plazentamembran abgeklemmt, da seine Struktur nahezu perfekt mit dem fetalen Gewebe übereinstimmt.
- Amniozentese. Bei der Untersuchung wird eine Fruchtwasserprobe entnommen. Dieses Verfahren erkennt das Edwards-Syndrom ab der 14. Schwangerschaftswoche.
- Cordozentese. Für die Analyse wird etwas fetales Nabelschnurblut benötigt, daher kommt diese Diagnosemethode ausschließlich in späteren Stadien ab der 20. Woche zum Einsatz.
Risiko eines Edwards-Syndroms laut Biochemie
Das pränatale Screening wird im ersten Schwangerschaftstrimester durchgeführt. Die werdende Mutter muss zwischen der 11. und 13. Schwangerschaftswoche Blut für die biochemische Analyse spenden. Basierend auf den Ergebnissen der Niveaubestimmung menschliches Choriongonadotropin und Plasmaprotein A wird das Risiko eines Edwards-Syndroms beim Fötus berechnet. Ist der Wert hoch, wird die Frau in die entsprechende Gruppe für die nächste Forschungsstufe (invasiv) aufgenommen.
Edwards-Syndrom – Anzeichen im Ultraschall
Diese Art der Diagnose wird selten verwendet, vor allem in Fällen, in denen die schwangere Frau keinem vorläufigen genetischen Screening unterzogen wurde. Das Edwards-Syndrom kann mittels Ultraschall erst in späteren Stadien erkannt werden, wenn der Fötus fast vollständig ausgebildet ist. Charakteristische Symptome Trisomie 18:
- intrauterine Defekte des Herz-Kreislauf- und Urogenitalsystems;
- Anomalien der Muskel-Skelett-Strukturen;
- Pathologien der Schädelknochen und der Weichteile des Kopfes.
- Indirekte Krankheitszeichen im Ultraschall:
- Bradykardie;
- verzögerte Entwicklung des Fötus;
- eine Arterie in der Nabelschnur (es sollten zwei sein);
- Hernie in der Bauchhöhle;
- visuelles Fehlen von Nasenknochen.
Edwards-Syndrom - Behandlung
Die Therapie der betrachteten Mutation zielt darauf ab, die Symptome zu lindern und das Leben des Babys zu erleichtern. Es ist unmöglich, das Edwards-Syndrom zu heilen und die volle Entwicklung des Kindes sicherzustellen. Medizinische Standardmaßnahmen helfen:
- Wiederherstellung der Nahrungspassage bei Anus- oder Darmatresie;
- die Ernährung durch einen Schlauch vor dem Hintergrund fehlender Saug- und Schluckreflexe organisieren;
- stabilisieren die Funktion des Herz-Kreislauf-Systems;
- normalisieren den Urinfluss.
Häufig erfordert das Edwards-Syndrom bei Neugeborenen zusätzlich den Einsatz entzündungshemmender, antibakterieller, hormoneller und anderer wirksamer Medikamente. Dies ist für die rechtzeitige intensive Behandlung aller dadurch hervorgerufenen Begleiterkrankungen erforderlich:
- Wilms-Tumor;
- Bindehautentzündung;
- Lungenentzündung;
- Mittelohrentzündung;
- pulmonale Hypertonie;
- Sinusitis;
- Urogenitalinfektionen;
- Stirnhöhlenentzündung;
- Bluthochdruck und mehr.
Edwards-Syndrom – Prognose
Die meisten Embryonen mit der beschriebenen genetischen Anomalie sterben während der Schwangerschaft aufgrund der Abstoßung des defekten Fötus durch den Körper. Auch nach der Geburt ist die Prognose enttäuschend. Wenn ein Edwards-Syndrom diagnostiziert wird, wie lange solche Kinder leben, berücksichtigen Sie in Prozent:
- 60 % – nicht länger als 3 Monate;
- 7-10 % – 1 Jahr;
- ca. 1 % – bis zu 10 Jahre.
In Ausnahmefällen (partielle oder mosaikartige Trisomie 18) können Einheiten die Reife erreichen. Selbst in solchen Situationen schreitet das John-Edwards-Syndrom unaufhaltsam voran. Erwachsene Kinder mit dieser Pathologie bleiben für immer oligophren. Das Maximum, das Sie ihnen beibringen können, ist:
- Kopf hoch;
- lächeln;
- iss alleine;
- einen begrenzten Personenkreis erkennen.
Edwards-Syndrom
Trisomie 18
ist eine schwere angeborene Erkrankung, die durch Chromosomenanomalien verursacht wird. Es ist eine der häufigsten Pathologien in dieser Kategorie (
Zweithäufigste Erkrankung nach dem Down-Syndrom
). Die Krankheit ist durch zahlreiche Störungen in der Entwicklung verschiedener Organe und Systeme gekennzeichnet. Die Prognose für das Kind ist meist ungünstig, doch hängt viel von der Betreuung durch die Eltern ab.
Die weltweite Prävalenz des Edwards-Syndroms schwankt zwischen 0,015 und 0,02 %. Es besteht keine eindeutige Abhängigkeit von Gebiet oder Rasse. Statistisch gesehen erkranken Mädchen drei- bis viermal häufiger als Jungen. Eine wissenschaftliche Erklärung für diesen Anteil konnte bislang nicht gefunden werden. Es wurde jedoch eine Reihe von Faktoren festgestellt, die das Risiko dieser Pathologie erhöhen können.
Wie andere Chromosomenmutationen ist das Edwards-Syndrom grundsätzlich eine unheilbare Krankheit. Modernste Behandlungs- und Betreuungsmethoden können das Kind nur am Leben erhalten und zu gewissen Fortschritten in seiner Entwicklung beitragen. Aufgrund der Vielzahl möglicher Erkrankungen und Komplikationen gibt es keine einheitlichen Empfehlungen für die Betreuung dieser Kinder.
Interessante Fakten
- Eine Beschreibung der Hauptsymptome dieser Krankheit erfolgte zu Beginn des 20. Jahrhunderts.
- Bis Mitte des 20. Jahrhunderts war es nicht möglich, ausreichende Informationen über diese Pathologie zu sammeln. Erstens erforderte dies eine entsprechende technologische Entwicklung, die den Nachweis des zusätzlichen Chromosoms ermöglichen würde. Zweitens starben die meisten Kinder bereits in den ersten Tagen oder Wochen ihres Lebens aufgrund der schlechten medizinischen Versorgung.
- Die erste vollständige Beschreibung der Krankheit und ihrer Hauptursache (das Auftreten eines zusätzlichen 18. Chromosoms) erfolgte erst 1960 durch den Arzt John Edward, nach dem die neue Pathologie damals benannt wurde.
- Die tatsächliche Inzidenz des Edwards-Syndroms liegt bei 1 Fall bei 2,5 bis 3.000 Empfängnissen (0,03 bis 0,04 %), die offiziellen Daten sind jedoch viel niedriger. Dies erklärt sich dadurch, dass fast die Hälfte der Embryonen mit dieser Anomalie nicht überleben und die Schwangerschaft mit einem Spontanabort oder dem intrauterinen Tod des Fötus endet. Eine detaillierte Diagnose der Ursache einer Fehlgeburt wird selten durchgeführt.
- Trisomie ist eine Variante einer Chromosomenmutation, bei der die Zellen einer Person nicht 46, sondern 47 Chromosomen enthalten. In dieser Krankheitsgruppe gibt es nur 3 Syndrome. Neben dem Edwards-Syndrom sind dies das Down-Syndrom (Trisomie 21) und das Patau-Syndrom (Trisomie 13). Bei Vorhandensein weiterer zusätzlicher Chromosomen ist die Pathologie nicht mit dem Leben vereinbar. Nur in diesen drei Fällen ist die Geburt eines lebenden Kindes und sein weiteres (wenn auch verlangsamtes) Wachstum und seine Entwicklung möglich.
Ursachen der genetischen Pathologie ist das Edwards-Syndrom Erbkrankheit, die durch das Vorhandensein eines zusätzlichen Chromosoms im menschlichen Genom gekennzeichnet ist. Um die Gründe zu verstehen, die die sichtbaren Manifestationen dieser Pathologie verursachen, ist es notwendig herauszufinden, was die Chromosomen selbst und das genetische Material als Ganzes sind.
Jede menschliche Zelle verfügt über einen Zellkern, der für die Speicherung und Verarbeitung genetischer Informationen verantwortlich ist. Der Kern enthält 46 Chromosomen (
), die ein mehrfach verpacktes Molekül sind
DNA-Desoxyribonukleinsäure
). Dieses Molekül enthält bestimmte Abschnitte, die Gene genannt werden. Jedes Gen ist der Prototyp eines bestimmten
im menschlichen Körper. Bei Bedarf liest die Zelle Informationen aus diesem Prototyp aus und produziert das entsprechende Protein. Gendefekte führen zur Produktion abnormaler Proteine, die für die Entstehung genetischer Krankheiten verantwortlich sind.
Ein Chromosomenpaar besteht aus zwei identischen DNA-Molekülen (
das eine ist väterlicherseits, das andere mütterlicherseits
), die durch eine kleine Brücke miteinander verbunden sind (
Zentromer
). Der Ort der Verbindung zweier Chromosomen eines Paares bestimmt die Form der gesamten Verbindung und ihr Aussehen unter dem Mikroskop.
Alle Chromosomen speichern unterschiedliche genetische Informationen (über verschiedene Proteine) und werden in folgende Gruppen eingeteilt:
- Gruppe A enthält 1 – 3 Chromosomenpaare, die groß und X-förmig sind;
- Gruppe B enthält 4–5 Chromosomenpaare, die ebenfalls groß sind, aber das Zentromer liegt weiter vom Zentrum entfernt, weshalb die Form dem Buchstaben X ähnelt, wobei das Zentrum nach unten oder oben verschoben ist;
- Gruppe C umfasst 6–12 Chromosomenpaare, die in ihrer Form den Chromosomen der Gruppe B ähneln, ihnen jedoch in der Größe unterlegen sind;
- Gruppe D umfasst 13 - 15 Chromosomenpaare, die sich durch mittlere Größe und die Lage des Zentromers ganz am Ende der Moleküle auszeichnen, was ihm eine Ähnlichkeit mit dem Buchstaben V verleiht;
- Gruppe E umfasst 16 - 18 Chromosomenpaare, die durch geringe Größe und mittlere Lage des Zentromers (X-förmig) gekennzeichnet sind;
- Gruppe F umfasst 19–20 Chromosomenpaare, die etwas kleiner als Chromosomen der Gruppe E und ähnlich geformt sind;
- Gruppe G umfasst 21–22 Chromosomenpaare, die sich durch eine V-Form und eine sehr geringe Größe auszeichnen.
Die oben genannten 22 Chromosomenpaare werden somatisch oder Autosomen genannt. Darüber hinaus gibt es Geschlechtschromosomen, die das 23. Paar bilden. Da sie sich im Aussehen nicht ähneln, wird jeder von ihnen separat bezeichnet. Das weibliche Geschlechtschromosom trägt die Bezeichnung Ist eines der Geschlechtschromosomen weiblich und das andere männlich, wird ein Junge geboren (Typ XY). Die Chromosomenformel wird Karyotyp genannt und kann wie folgt bezeichnet werden: 46,XX. Hier bezeichnet die Zahl 46 die Gesamtzahl der Chromosomen (23 Paare) und XX ist die Formel der Geschlechtschromosomen, die vom Geschlecht abhängt (das Beispiel zeigt den Karyotyp einer normalen Frau).
Unter dem Edwards-Syndrom versteht man sogenannte Chromosomenerkrankungen, bei denen es sich nicht um einen Gendefekt, sondern um einen Defekt im gesamten DNA-Molekül handelt. Genauer gesagt handelt es sich bei der klassischen Form dieser Krankheit um das Vorhandensein eines zusätzlichen 18. Chromosoms. Der Karyotyp wird in solchen Fällen als 47,XX, 18+ (
für Mädchen
) und 47,ХY, 18+ (
für Junge
). Die letzte Ziffer gibt die Nummer des zusätzlichen Chromosoms an. Überschüssige genetische Informationen in Zellen führen zum Auftreten entsprechender Krankheitserscheinungen, die zusammenfassend als „Edwards-Syndrom“ bezeichnet werden. Verfügbarkeit zusätzlicher (
) Chromosom Nummer 18 ergab ein weiteres (
wissenschaftlicher
) Die Krankheit heißt Trisomie 18.
Abhängig von der Form des Chromosomendefekts werden drei Arten dieser Erkrankung unterschieden:
- Vollständige Trisomie 18. Bei der vollständigen oder klassischen Form des Edwards-Syndroms verfügen alle Zellen im Körper über ein zusätzliches Chromosom. Diese Krankheitsvariante kommt in über 90 % der Fälle vor und ist die schwerste.
- Partielle Trisomie 18. Die partielle Trisomie 18 ist ein sehr seltenes Phänomen (nicht mehr als 3 % aller Fälle des Edwards-Syndroms). Damit enthalten die Körperzellen nicht ein ganzes zusätzliches Chromosom, sondern nur einen Bruchteil davon. Dieser Defekt kann die Folge einer unsachgemäßen Aufteilung des genetischen Materials sein, kommt aber sehr selten vor. Manchmal bindet ein Teil des achtzehnten Chromosoms an ein anderes DNA-Molekül (wird in seine Struktur eingefügt, wodurch das Molekül verlängert wird, oder „klammert“ sich einfach mit Hilfe einer Brücke). Die anschließende Zellteilung führt dazu, dass der Körper über zwei normale Chromosomen mit der Nummer 18 und einige weitere Gene dieser Chromosomen (ein konserviertes Fragment des DNA-Moleküls) verfügt. In diesem Fall wird die Zahl der Geburtsfehler deutlich geringer sein. Es gibt nicht die gesamte im 18. Chromosom kodierte genetische Information, sondern nur einen Teil davon. Patienten mit partieller Trisomie 18 haben eine bessere Prognose als Kinder mit der Vollform, bleiben aber weiterhin schlecht.
- Mosaikform. Die Mosaikform des Edwards-Syndroms tritt in 5–7 % der Fälle dieser Erkrankung auf. Der Mechanismus seines Auftretens unterscheidet sich von anderen Arten. Tatsache ist, dass hier der Defekt nach der Verschmelzung von Sperma und Ei entstanden ist. Beide Gameten (Geschlechtszellen) hatten zunächst einen normalen Karyotyp und trugen ein Chromosom jedes Typs. Nach der Fusion entstand eine Zelle mit der normalen Formel 46,XX oder 46,XY. Bei der Teilung dieser Zelle ist eine Fehlfunktion aufgetreten. Bei der Verdoppelung des genetischen Materials erhielt eines der Fragmente ein zusätzliches 18. Chromosom. So hat sich in einem bestimmten Stadium ein Embryo gebildet, dessen Zellen teilweise einen normalen Karyotyp haben (zum Beispiel 46,XX) und andere den Karyotyp des Edwards-Syndroms (47,XX, 18+). Der Anteil pathologischer Zellen übersteigt nie 50 %. Ihre Anzahl hängt davon ab, in welchem Teilungsstadium der Ausgangszelle der Fehler aufgetreten ist. Je später dies geschieht, desto geringer ist der Anteil defekter Zellen. Die Form erhielt ihren Namen aufgrund der Tatsache, dass alle Zellen des Körpers eine Art Mosaik darstellen. Einige von ihnen sind gesund, andere haben eine schwere genetische Pathologie. In diesem Fall gibt es kein Muster in der Verteilung der Zellen im Körper, das heißt, alle defekten Zellen können nicht an nur einer Stelle lokalisiert werden, um sie entfernen zu können. Der Allgemeinzustand des Patienten ist einfacher als bei der klassischen Form der Trisomie 18.
Das Vorhandensein eines zusätzlichen Chromosoms im menschlichen Genom bringt viele Probleme mit sich. Tatsache ist, dass menschliche Zellen darauf programmiert sind, genetische Informationen zu lesen und nur die von der Natur vorgegebene Anzahl an DNA-Molekülen zu duplizieren. Störungen bereits in der Struktur eines Gens können zu schweren Krankheiten führen. Bei Vorhandensein eines vollständigen DNA-Moleküls kommt es bereits im Stadium der intrauterinen Entwicklung vor der Geburt des Kindes zu multiplen Störungen.
Jüngsten Forschungsergebnissen zufolge enthält das Chromosom Nummer 18 557 Gene, die für mindestens 289 verschiedene Proteine kodieren. Prozentual sind das etwa 2,5 % des gesamten Erbguts. Die Störungen, die ein so großes Ungleichgewicht verursacht, sind sehr schwerwiegend. Eine falsche Menge an Proteinen führt zu vielen Anomalien in der Entwicklung verschiedener Organe und Gewebe. Beim Edwards-Syndrom sind am häufigsten die Schädelknochen, einige Teile des Nervensystems, das Herz-Kreislauf- und Urogenitalsystem betroffen. Dies liegt offenbar daran, dass die auf diesem Chromosom befindlichen Gene mit der Entwicklung dieser bestimmten Organe und Systeme zusammenhängen.
Somit ist die Haupt- und einzige Ursache des Edwards-Syndroms das Vorhandensein eines zusätzlichen DNA-Moleküls. Meistens (
in der klassischen Form der Erkrankung
) wird von einem der Eltern geerbt. Normalerweise ist jeder Gamete (
Sperma und Ei
) enthalten 22 ungepaarte somatische Chromosomen sowie ein Geschlechtschromosom. Eine Frau gibt dem Kind immer den Standardsatz 22+X weiter, und ein Mann kann 22+X oder 22+Y weitergeben. Dadurch wird das Geschlecht des Kindes bestimmt. Die Geschlechtszellen der Eltern entstehen durch die Teilung gewöhnlicher Zellen in zwei Gruppen. Normalerweise ist die Mutterzelle in zwei gleiche Teile geteilt, manchmal sind jedoch nicht alle Chromosomen in zwei Hälften geteilt. Wenn sich das 18. Paar nicht an den Polen der Zelle getrennt hat, dann ist eines der Eier (
oder eines der Spermien
) wird von vornherein defekt sein. Es wird nicht 23, sondern 24 Chromosomen haben. Wenn diese Zelle an der Befruchtung beteiligt ist, erhält das Kind ein zusätzliches 18. Chromosom.
Die folgenden Faktoren können eine fehlerhafte Zellteilung beeinflussen:
- Alter der Eltern. Es ist erwiesen, dass die Wahrscheinlichkeit von Chromosomenanomalien direkt proportional zum Alter der Mutter steigt. Beim Edwards-Syndrom ist dieser Zusammenhang weniger ausgeprägt als bei anderen ähnlichen Pathologien (z. B. Down-Syndrom). Bei Frauen über 40 Jahren ist das Risiko, ein Kind mit dieser Pathologie zu bekommen, jedoch im Durchschnitt sechs- bis siebenmal höher. Diese Abhängigkeit vom Alter des Vaters ist in deutlich geringerem Maße zu beobachten.
- Rauchen und Alkohol. Schlechte Gewohnheiten wie Rauchen und Alkoholmissbrauch können das menschliche Fortpflanzungssystem beeinträchtigen und die Teilung der Keimzellen beeinträchtigen. Daher erhöht der regelmäßige Konsum dieser Substanzen (wie auch anderer Medikamente) das Risiko einer unsachgemäßen Verteilung des genetischen Materials.
- Einnahme von Medikamenten. Einige Medikamente können bei falscher Einnahme im ersten Trimester die Teilung der Keimzellen beeinträchtigen und die Mosaikform des Edwards-Syndroms hervorrufen.
- Erkrankungen des Genitalbereichs. Frühere Infektionen der Fortpflanzungsorgane können die ordnungsgemäße Zellteilung beeinträchtigen. Sie erhöhen das Risiko für chromosomale und genetische Erkrankungen im Allgemeinen, obwohl ähnliche Studien speziell für das Edwards-Syndrom nicht durchgeführt wurden.
- Strahlung. Die Einwirkung von Röntgenstrahlen oder anderer ionisierender Strahlung auf die Genitalien kann zu genetischen Mutationen führen. Besonders gefährlich sind solche äußeren Einflüsse im Jugendalter, wenn die Zellteilung am aktivsten erfolgt. Die Partikel, aus denen die Strahlung besteht, dringen leicht in das Gewebe ein und setzen das DNA-Molekül einer Art „Bombardierung“ aus. Geschieht dies zum Zeitpunkt der Zellteilung, ist das Risiko einer Chromosomenmutation besonders hoch.
Im Allgemeinen kann man nicht sagen, dass die Ursachen für die Entstehung des Edwards-Syndroms vollständig bekannt und gut untersucht sind. Die oben genannten Faktoren erhöhen nur das Risiko, diese Mutation zu entwickeln. Es ist auch möglich, dass manche Menschen eine angeborene Veranlagung für eine falsche Verteilung des genetischen Materials in den Keimzellen haben. Man geht beispielsweise davon aus, dass bei einem Ehepaar, das bereits ein Kind mit Edwards-Syndrom zur Welt gebracht hat, die Wahrscheinlichkeit, ein zweites Kind mit einer ähnlichen Pathologie zu bekommen, bei bis zu 2–3 % liegt (etwa 200-mal höher als der Durchschnitt). Prävalenz dieser Krankheit).
Wie sehen Neugeborene mit Edwards-Syndrom aus?
Wie Sie wissen, kann das Edwards-Syndrom bereits vor der Geburt diagnostiziert werden, in den meisten Fällen wird diese Krankheit jedoch unmittelbar nach der Geburt des Kindes erkannt. Neugeborene mit dieser Pathologie weisen eine Reihe ausgeprägter Entwicklungsanomalien auf, die manchmal sofort den Verdacht auf die richtige Diagnose zulassen. Die Bestätigung erfolgt später durch eine spezielle genetische Analyse.
Neugeborene mit Edwards-Syndrom weisen folgende charakteristische Entwicklungsstörungen auf:
- Veränderung der Schädelform;
- Veränderung der Ohrenform;
- Anomalien der Gaumenentwicklung;
- Wippenfuß;
- abnormale Fingerlänge;
- Veränderung der Form des Unterkiefers;
- Verschmelzung der Finger;
- abnormale Entwicklung der Geschlechtsorgane;
- Beugestellung der Hände;
- dermatoglyphische Zeichen.
Veränderungen der Schädelform Ein typisches Symptom des Edwards-Syndroms ist die Dolichozephalie. Dies ist die Bezeichnung für eine charakteristische Veränderung der Kopfform eines Neugeborenen, die auch bei einigen anderen genetisch bedingten Erkrankungen auftritt. Dolichocephals (Kinder mit diesem Symptom) haben einen längeren und schmaleren Schädel. Das Vorliegen dieser Anomalie wird durch spezielle Messungen genau bestätigt. Bestimmt wird das Verhältnis der Schädelbreite auf Höhe der Scheitelknochen zur Schädellänge (vom Vorsprung über dem Nasenrücken bis zum Hinterhauptsvorsprung). Wenn das resultierende Verhältnis weniger als 75 % beträgt, handelt es sich bei dem Kind um einen Dolichozephalen. Dieses Symptom an sich ist keine ernsthafte Störung. Dies ist nur eine Form der Schädelform, die auch bei ganz normalen Menschen vorkommt. Bei Kindern mit Edwards-Syndrom handelt es sich in 80 - 85 % der Fälle um ausgeprägte Dolichocephalen, bei denen ohne besondere Messungen das Missverhältnis von Schädellänge und -breite auffällt.
Eine weitere Variante der Fehlentwicklung des Schädels ist die sogenannte Mikrozephalie, bei der der Kopf insgesamt im Vergleich zum Rest des Körpers zu klein ist. Dies gilt zunächst nicht für den Gesichtsschädel (
Kiefer, Wangenknochen, Augenhöhlen
), nämlich der Schädel, in dem sich das Gehirn befindet. Mikrozephalie kommt beim Edwards-Syndrom seltener vor als Dolichozephalie, kommt aber auch häufiger vor als bei gesunden Menschen.
Die Form des Ohrs verändern
Wenn Dolichozephalie eine normale Variante sein kann, ist die Pathologie der Entwicklung der Ohrmuschel bei Kindern mit Edwards-Syndrom viel schwerwiegender. Bis zu einem gewissen Grad wird dieses Symptom bei mehr als 95 % der Kinder mit der Vollform dieser Krankheit beobachtet. In Mosaikform ist seine Häufigkeit etwas geringer. Die Ohrmuschel liegt normalerweise tiefer als bei normalen Menschen (
manchmal unter Augenhöhe
). Die charakteristischen Höcker des Knorpels, der die Ohrmuschel bildet, sind schlecht definiert oder fehlen. Der Lappen oder Tragus kann auch fehlen (
ein kleiner hervorstehender Knorpelbereich vor der Gehöröffnung
). Der Gehörgang selbst ist meist verengt und fehlt in etwa 20–25 % vollständig.
Anomalien in der Entwicklung des Gaumens Die Gaumenfortsätze des Oberkiefers verschmelzen während der Embryonalentwicklung zum harten Gaumen. Bei Kindern mit Edwards-Syndrom bleibt dieser Prozess oft unvollständig. An der Stelle, an der sich bei normalen Menschen die Mittelnaht befindet (sie ist mit der Zunge in der Mitte des harten Gaumens zu ertasten), haben sie einen Längsriss.
Es gibt mehrere Varianten dieses Defekts:
- Gaumenspalte (der hintere, tiefe Teil des Gaumens, der über dem Rachen hängt);
- Teilspalte des harten Gaumens (die Lücke erstreckt sich nicht über den gesamten Oberkiefer);
- völlige Nichtverschmelzung des harten und weichen Gaumens;
- völlige Nichtverschmelzung von Gaumen und Lippe.
In einigen Fällen ist die Gaumenspalte beidseitig. Die beiden nach oben ragenden Ecken der Oberlippe sind der Beginn pathologischer Risse. Aufgrund dieses Defekts kann das Kind seinen Mund nicht vollständig schließen. In schweren Fällen ist die Kommunikation zwischen Mund- und Nasenhöhle deutlich sichtbar (auch bei geschlossenem Mund). Vorderzähne können in Zukunft fehlen oder zur Seite wachsen.
Diese Entwicklungsstörungen werden auch als Gaumenspalte, Gaumenspalte und Lippenspalte bezeichnet. Alle von ihnen können außerhalb des Edwards-Syndroms auftreten, aber bei Kindern mit dieser Pathologie ist ihre Häufigkeit besonders hoch (
fast 20 % der Neugeborenen
). Viel häufiger (
bis zu 65 % der Neugeborenen
) haben ein weiteres Merkmal, das als hoher oder gotischer Himmel bekannt ist. Sie kann als normale Variante eingestuft werden, da sie auch bei gesunden Menschen auftritt.
Das Vorhandensein einer Gaumen- oder Oberlippenspalte ist kein Beweis für das Edwards-Syndrom. Diese Fehlbildung kann mit relativ hoher Häufigkeit und unabhängig voneinander ohne Begleitstörungen anderer Organe und Systeme auftreten. Um diese Anomalie zu korrigieren, gibt es eine Reihe standardmäßiger chirurgischer Eingriffe.
Schaukelfuß
So bezeichnet man eine charakteristische Fußveränderung, die vor allem im Rahmen des Edwards-Syndroms auftritt. Die Häufigkeit bei dieser Krankheit beträgt 75 %. Der Defekt besteht in einer falschen Beziehung zwischen Talus, Fersenbein und Strahlbein. Bei Kindern wird sie als Plattvalgusfußdeformität klassifiziert.
Äußerlich sieht der Fuß eines Neugeborenen so aus. Der Fersenhöcker, auf dem der Fußrücken aufliegt, ragt nach hinten. Der Bogen kann vollständig fehlen. Dies ist leicht zu erkennen, wenn man den Fuß von innen betrachtet. Normalerweise erscheint dort eine konkave Linie, die von der Ferse bis zur Basis des großen Zehs verläuft. Bei einem Kippfuß gibt es diese Linie nicht. Der Fuß ist flach oder sogar konvex. Dadurch sieht es aus wie die Beine eines Schaukelstuhls.
Abnormale Fingerlänge
Kinder mit Edwards-Syndrom können aufgrund von Veränderungen in der Struktur ihrer Füße abnormale Längenproportionen ihrer Zehen aufweisen. Dabei handelt es sich insbesondere um den Daumen, der normalerweise der längste ist. Bei Neugeborenen mit diesem Syndrom ist die Länge geringer als der zweite Finger. Dieser Defekt kann nur bemerkt werden, wenn die Finger gestreckt und sorgfältig untersucht werden. Mit zunehmendem Alter, wenn das Kind wächst, wird es deutlicher. Da die Verkürzung des Hallux vor allem bei Kippfüßen auftritt, ist die Prävalenz dieser Symptome bei Neugeborenen ungefähr gleich.
Bei Erwachsenen hat eine Verkürzung der Großzehe nicht den gleichen diagnostischen Wert. Ein solcher Defekt kann ein individuelles Merkmal eines gesunden Menschen oder eine Folge des Einflusses anderer Faktoren sein (
Verformung der Gelenke, Knochenerkrankungen, Tragen von Schuhen, die nicht richtig passen
). In diesem Zusammenhang sollte dieses Zeichen nur bei Neugeborenen als mögliches Symptom in Betracht gezogen werden, wenn andere Entwicklungsanomalien vorliegen.
Veränderung der Form des Unterkiefers
Formveränderungen des Unterkiefers treten bei Neugeborenen in fast 70 % der Fälle auf. Normalerweise ragt das Kinn bei Kindern nicht so stark hervor wie bei Erwachsenen, bei Patienten mit Edwards-Syndrom ist es jedoch zu stark zurückgezogen. Dies geschieht aufgrund einer Unterentwicklung des Unterkiefers, die als Mikrognathie bezeichnet wird (
Mikrogenie
). Dieses Symptom tritt auch bei anderen angeborenen Erkrankungen auf. Es ist nicht so selten, Erwachsene mit ähnlichen Gesichtszügen zu finden. In Ermangelung begleitender Pathologien gilt dies als Variante der Norm, führt jedoch zu einigen Schwierigkeiten.
Neugeborene mit Mikrognathie entwickeln in der Regel schnell folgende Probleme:
- Unfähigkeit, den Mund über längere Zeit geschlossen zu halten (Speichelaustritt);
- Schwierigkeiten beim Füttern;
- späte Entwicklung der Zähne und deren falsche Lage.
Der Abstand zwischen Unter- und Oberkiefer kann mehr als 1 cm betragen, was angesichts der Kopfgröße des Babys sehr groß ist.
Fingerfusion
Bei etwa 45 % der Neugeborenen wird eine Verschmelzung der Finger oder wissenschaftlich Syndaktylie beobachtet. Am häufigsten betrifft diese Anomalie die Zehen, Syndaktylie tritt jedoch auch an den Händen auf. In milden Fällen wird die Fusion durch eine Hautfalte wie eine kurze Membran gebildet. In schwereren Fällen wird eine Fusion von Knochengewebe durch Brücken beobachtet.
Syndaktylie kommt nicht nur beim Edwards-Syndrom vor, sondern auch bei vielen anderen Chromosomenerkrankungen. Es gibt auch Fälle, in denen dieser Entwicklungsfehler der einzige war und sich der Patient ansonsten nicht von normalen Kindern unterschied. In dieser Hinsicht ist die Fingerfusion nur eines der möglichen Anzeichen des Edwards-Syndroms, das zur Vermutung der Diagnose beiträgt, diese jedoch nicht bestätigt.
Anomalien in der Entwicklung der Geschlechtsorgane
Gleich nach
Bei Neugeborenen mit Edwards-Syndrom kann manchmal eine abnormale Entwicklung der äußeren Genitalien beobachtet werden. Sie gehen in der Regel mit Entwicklungsstörungen des gesamten Urogenitalapparates einher, die jedoch ohne spezielle diagnostische Maßnahmen nicht festgestellt werden können. Die häufigsten äußerlich erkennbaren Anomalien sind eine Unterentwicklung des Penis bei Jungen und eine Hypertrophie (
an Größe zunehmen
) Klitoris bei Mädchen. Sie treten in etwa 15–20 % der Fälle auf. Etwas seltener kann eine abnormale Lage der Harnröhre beobachtet werden (
Hypospadie
) oder Fehlen von Hoden im Hodensack bei Jungen (
Kryptorchismus
Beugehaltung der Hände
Bei der Beugestellung der Hände handelt es sich um eine besondere Anordnung der Finger, die weniger auf Strukturstörungen im Handbereich als vielmehr auf einen erhöhten Muskeltonus zurückzuführen ist. Die Beugemuskeln der Finger und der Hand sind ständig angespannt, weshalb Daumen und kleiner Finger scheinbar die anderen Finger bedecken, die gegen die Handfläche gedrückt werden. Dieses Symptom wird bei vielen angeborenen Pathologien beobachtet und ist nicht spezifisch für das Edwards-Syndrom. Wenn jedoch ein Pinsel dieser Form entdeckt wird, muss von dieser Pathologie ausgegangen werden. Damit wird bei fast 90 % der Neugeborenen eine Beugestellung der Finger beobachtet.
Dermatoglyphische Zeichen Viele Chromosomenanomalien bei Neugeborenen weisen charakteristische dermatoglyphische Veränderungen auf (abnormale Muster und Falten auf der Haut der Handflächen). Beim Edwards-Syndrom können in fast 60 % der Fälle einige Anzeichen festgestellt werden. Sie sind vor allem für die vorläufige Diagnose von Mosaik- oder Teilformen der Erkrankung wichtig. Bei vollständiger Trisomie 18 wird nicht auf Dermatoglyphen zurückgegriffen, da andere, auffälligere Entwicklungsanomalien ausreichen, um ein Edwards-Syndrom zu vermuten.
Die wichtigsten dermatoglyphischen Zeichen des Edwards-Syndroms sind:
- Bögen an den Fingerspitzen sind häufiger zu finden als bei gesunden Menschen;
- die Hautfalte zwischen dem letzten (Nagel) und dem vorletzten (mittleren) Fingerglied fehlt;
- 30 % der Neugeborenen haben eine sogenannte Querfurche an der Handfläche (Affenfalte, Affenfalte).
Spezielle Untersuchungen können andere Abweichungen von der Norm aufdecken, aber unmittelbar nach der Geburt, ohne Einbeziehung spezialisierter Spezialisten, reichen diese Veränderungen für Ärzte aus.
Zusätzlich zu den oben genannten Anzeichen gibt es eine Reihe möglicher Entwicklungsanomalien, die bei der vorläufigen Diagnose des Edwards-Syndroms hilfreich sein können. Einigen Daten zufolge kann eine detaillierte äußere Untersuchung bis zu 50 äußere Anzeichen erkennen. Die Kombination der oben dargestellten häufigsten Symptome weist höchstwahrscheinlich darauf hin, dass das Kind an dieser schwerwiegenden Pathologie leidet. Bei der mosaikartigen Variante des Edwards-Syndroms kann es zwar nicht zu mehreren Anomalien kommen, aber schon das Vorliegen einer davon ist ein Hinweis auf einen speziellen Gentest.
Wie sehen Kinder mit Edwards-Syndrom aus? Kinder mit Edwards-Syndrom entwickeln mit zunehmendem Alter typischerweise eine Vielzahl von Begleiterkrankungen. Ihre Symptome treten bereits wenige Wochen nach der Geburt auf. Diese Symptome können die erste Manifestation des Syndroms sein, da bei der Mosaikvariante in seltenen Fällen die Krankheit unmittelbar nach der Geburt unbemerkt bleiben kann. Dann wird die Diagnose der Krankheit komplizierter.
Die meisten der bei der Geburt festgestellten äußeren Erscheinungsformen des Syndroms bleiben bestehen und machen sich deutlicher bemerkbar. Wir sprechen über die Form des Schädels, den Kippfuß, die Verformung der Ohrmuschel usw. Allmählich kommen weitere äußere Erscheinungen hinzu, die unmittelbar nach der Geburt nicht bemerkt werden konnten. In diesem Fall handelt es sich um Anzeichen, die bei Kindern im ersten Lebensjahr auftreten können.
Kinder mit Edwards-Syndrom weisen folgende äußere Merkmale auf:
- Verzögerung der körperlichen Entwicklung;
- Klumpfuß;
- abnormaler Muskeltonus;
- abnormale emotionale Reaktionen.
Eine verzögerte körperliche Entwicklung wird durch das niedrige Geburtsgewicht des Kindes erklärt (nur 2000–2200 g im normalen Gestationsalter). Eine wesentliche Rolle spielt auch ein genetischer Defekt, der nicht dazu führt, dass sich alle Körpersysteme normal und harmonisch entwickeln können. Die Hauptindikatoren, anhand derer das Wachstum und die Entwicklung eines Kindes beurteilt werden, sind stark reduziert.
Sie können die Verzögerung eines Kindes anhand der folgenden anthropometrischen Indikatoren erkennen:
- Größe des Kindes;
- Gewicht des Kindes;
- Brustumfang;
- Kopfumfang (dieser Indikator kann normal oder sogar erhöht sein, ist jedoch aufgrund einer angeborenen Verformung des Schädels nicht verlässlich).
Klumpfuß Klumpfuß ist eine Folge einer Verformung der Knochen und Gelenke der Füße sowie einer mangelnden normalen Kontrolle durch das Nervensystem. Kinder haben Schwierigkeiten, mit dem Laufen zu beginnen (die meisten erreichen dieses Stadium aufgrund angeborener Fehlbildungen nicht). Äußerlich kann das Vorliegen eines Klumpfußes anhand der Verformung der Füße und der abnormalen Ruhestellung der Beine beurteilt werden.
Anormaler Muskeltonus
Ein abnormaler Tonus, der bei der Geburt zu einer Beugestellung der Hand führt, beginnt mit dem Wachstum auch in anderen Muskelgruppen aufzutreten. Am häufigsten haben Kinder mit Edwards-Syndrom eine verminderte Muskelkraft, sie sind schlaff und haben keinen normalen Tonus. Abhängig von der Art der Schädigung des Zentralnervensystems kann es bei einigen Gruppen zu einem erhöhten Tonus kommen, der sich in spastischen Kontraktionen dieser Muskeln äußert (
zum Beispiel Armbeuger oder Beinstrecker
). Äußerlich äußert sich dies durch einen Mangel an minimaler Bewegungskoordination. Manchmal führen spastische Kontraktionen zu abnormalen Beugungen der Gliedmaßen oder sogar zu Luxationen.
Abnormale emotionale Reaktionen
Das Fehlen oder der abnormale Ausdruck jeglicher Emotionen ist eine Folge von Anomalien in der Entwicklung bestimmter Teile des Gehirns (
am häufigsten das Kleinhirn und das Corpus callosum
). Diese Veränderungen führen zu einer schweren geistigen Behinderung, die ausnahmslos bei allen Kindern mit Edwards-Syndrom beobachtet wird. Äußerlich äußert sich ein niedriger Entwicklungsstand durch einen charakteristischen „fehlenden“ Gesichtsausdruck und mangelnde emotionale Reaktion auf äußere Reize. Das Kind hält den Blickkontakt nicht gut (
folgt nicht der Bewegung des Fingers vor den Augen usw.
). Eine mangelnde Reaktion auf scharfe Geräusche kann eine Folge einer Schädigung sowohl des Nervensystems als auch des Hörgeräts sein. All diese Anzeichen zeigen sich, wenn das Kind in den ersten Lebensmonaten heranwächst.
Wie sehen Erwachsene mit Edwards-Syndrom aus?
In den allermeisten Fällen erreichen Kinder, die mit dem Edwards-Syndrom geboren werden, nicht das Erwachsenenalter. In der Vollform dieser Krankheit, wenn in jeder Körperzelle ein zusätzliches Chromosom vorhanden ist, sterben 90 % der Kinder vor dem Alter von einem Jahr aufgrund schwerwiegender Anomalien in der Entwicklung innerer Organe. Auch bei chirurgischer Korrektur möglicher Defekte und hochwertiger Pflege ist ihr Körper anfälliger für Infektionskrankheiten. Begünstigt wird dies auch durch Essstörungen, die bei den meisten Kindern auftreten. All dies erklärt die höchste Sterblichkeitsrate beim Edwards-Syndrom.
Bei einer milderen Mosaikform, bei der nur ein Teil der Zellen im Körper einen abnormalen Chromosomensatz enthält, ist die Überlebensrate etwas höher. Allerdings erreichen auch in diesen Fällen nur wenige Patienten das Erwachsenenalter. Ihr Aussehen wird durch angeborene Anomalien bestimmt, die bei der Geburt vorhanden waren (
Lippenspalte, deformiertes Ohr usw.
). Das Hauptsymptom, das ausnahmslos bei allen Kindern auftritt, ist eine schwere geistige Behinderung. Wenn ein Kind mit Edwards-Syndrom das Erwachsenenalter erreicht hat, ist es zutiefst geistig zurückgeblieben (
IQ unter 20, was dem schwersten Grad geistiger Behinderung entspricht
). Generell werden in der medizinischen Fachliteratur Einzelfälle beschrieben, bei denen Kinder mit Edwards-Syndrom das Erwachsenenalter erreichten. Aus diesem Grund liegen zu wenig objektive Daten vor, um über die äußeren Anzeichen dieser Krankheit bei Erwachsenen zu sprechen.
Diagnose genetischer Pathologie
Derzeit gibt es drei Hauptstadien bei der Diagnose des Edwards-Syndroms, die jeweils mehrere mögliche Methoden umfassen. Da diese Krankheit unheilbar ist, sollten Eltern auf die Möglichkeiten dieser Methoden achten und diese nutzen. Die meisten Tests werden in speziellen Pränataldiagnostikzentren durchgeführt, in denen alle notwendigen Geräte zur Suche nach genetisch bedingten Erkrankungen vorhanden sind. Allerdings kann auch eine Beratung durch einen Genetiker oder Neonatologen sinnvoll sein.
Die Diagnose des Edwards-Syndroms ist in folgenden Stadien möglich:
- Diagnose vor der Empfängnis;
- Diagnostik während der intrauterinen Entwicklung;
- Diagnose nach der Geburt.
Diagnostik vor der Empfängnis Die Diagnostik vor der Empfängnis eines Kindes ist eine ideale Option, aber leider sind ihre Möglichkeiten im gegenwärtigen Entwicklungsstadium der Medizin sehr begrenzt. Ärzte können mehrere Methoden anwenden, um auf eine erhöhte Wahrscheinlichkeit hinzuweisen, ein Kind mit einer Chromosomenstörung zu bekommen, aber nichts weiter. Tatsache ist, dass beim Edwards-Syndrom grundsätzlich keine Störungen bei den Eltern festgestellt werden können. Eine defekte Keimzelle mit 24 Chromosomen ist nur eine von vielen Tausenden. Deshalb kann man vor dem Zeitpunkt der Empfängnis nicht mit Sicherheit sagen, ob ein Kind mit dieser Krankheit zur Welt kommt.
Die wichtigsten diagnostischen Methoden vor der Empfängnis sind:
- Familiengeschichte. Eine Familienanamnese ist eine detaillierte Befragung beider Elternteile zu ihrer Abstammung. Der Arzt interessiert sich für alle Fälle von erblichen (insbesondere chromosomalen) Erkrankungen in der Familie. Wenn sich mindestens ein Elternteil an einen Fall von Trisomie (Edwards-, Down-, Patau-Syndrom) erinnert, erhöht sich die Wahrscheinlichkeit, ein erkranktes Kind zu bekommen, erheblich. Das Risiko beträgt jedoch immer noch nicht mehr als 1 %. Bei wiederholten Fällen dieser Erkrankungen bei Vorfahren steigt das Risiko um ein Vielfaches. Im Wesentlichen läuft die Analyse auf eine Konsultation mit einem Neonatologen oder Genetiker hinaus. Eltern können im Vorfeld versuchen, detailliertere Informationen über ihre Vorfahren (vorzugsweise 3 bis 4 Generationen) zu sammeln. Dadurch wird die Genauigkeit dieser Methode erhöht.
- Erkennung von Risikofaktoren. Der Hauptrisikofaktor, der das Risiko für Chromosomenanomalien objektiv erhöht, ist das Alter der Mutter. Wie oben erwähnt, steigt für Mütter ab dem 40. Lebensjahr die Wahrscheinlichkeit, ein Kind mit Edwards-Syndrom zu bekommen, um ein Vielfaches. Einigen Daten zufolge geht nach 45 Jahren (Alter der Mutter) fast jede fünfte Schwangerschaft mit einer Chromosomenpathologie einher. Die meisten davon enden mit einer Fehlgeburt. Weitere Faktoren sind frühere Infektionskrankheiten, chronische Krankheiten und schlechte Gewohnheiten. Ihre Rolle bei der Diagnose ist jedoch viel geringer. Auch auf die Frage, ob ein Kind mit Edwards-Syndrom gezeugt wird, gibt diese Methode keine genaue Antwort.
- Genetische Analyse der Eltern. Beschränkten sich frühere Methoden auf die Befragung der Eltern, handelt es sich bei der Genanalyse um eine vollwertige Studie, die spezielle Geräte, Reagenzien und qualifizierte Fachkräfte erfordert. Den Eltern wird Blut entnommen, aus dem im Labor Leukozyten isoliert werden. Nach der Behandlung mit speziellen Substanzen werden in diesen Zellen Chromosomen im Teilungsstadium deutlich sichtbar. Auf diese Weise wird der Karyotyp der Eltern ermittelt. In den allermeisten Fällen ist es normal (bei hier nachweisbaren Chromosomenanomalien ist die Wahrscheinlichkeit einer Fortpflanzung vernachlässigbar gering). Darüber hinaus ist es mithilfe spezieller Marker (Fragmente von Molekülketten) möglich, DNA-Abschnitte mit defekten Genen zu erkennen. Hierbei handelt es sich jedoch nicht um Chromosomenanomalien, sondern um genetische Mutationen, die keinen direkten Einfluss auf die Wahrscheinlichkeit eines Edwards-Syndroms haben. Daher liefert die genetische Analyse der Eltern vor der Empfängnis trotz der Komplexität und der hohen Kosten auch keine eindeutige Antwort auf die Prognose dieser Pathologie.
Diagnostik während der fetalen Entwicklung Während der intrauterinen Entwicklung gibt es mehrere Methoden, die das Vorliegen einer Chromosomenpathologie beim Fötus direkt oder indirekt bestätigen können. Die Genauigkeit dieser Methoden ist viel höher, da sich Ärzte nicht mit den Eltern, sondern mit dem Fötus selbst befassen. Zur Untersuchung stehen sowohl der Embryo selbst als auch seine Zellen mit eigener DNA zur Verfügung. Dieses Stadium wird auch Pränataldiagnostik genannt und ist das wichtigste. Zu diesem Zeitpunkt können Sie die Diagnose bestätigen, die Eltern vor dem Vorliegen einer Pathologie warnen und gegebenenfalls die Schwangerschaft abbrechen. Wenn sich eine Frau für die Geburt entscheidet und das Neugeborene noch am Leben ist, haben die Ärzte die Möglichkeit, sich im Voraus darauf vorzubereiten, ihm die notwendige Pflege zukommen zu lassen.
Die wichtigsten Forschungsmethoden im Rahmen der Pränataldiagnostik sind:
- Ultraschalluntersuchung (Ultraschall). Diese Methode ist nicht-invasiv, das heißt, das Gewebe der Mutter oder des Fötus wird nicht geschädigt. Es ist absolut sicher und wird im Rahmen der Pränataldiagnostik allen schwangeren Frauen empfohlen (unabhängig von ihrem Alter oder einem erhöhten Risiko für Chromosomenerkrankungen). Das Standardprogramm geht davon aus, dass Ultraschall dreimal durchgeführt werden sollte (in der 10.–14., 20.–24. und 32.–34. Schwangerschaftswoche). Wenn der behandelnde Arzt die Möglichkeit angeborener Fehlbildungen vermutet, können außerplanmäßige Ultraschalluntersuchungen durchgeführt werden. Das Edwards-Syndrom kann durch eine Verzögerung der Größe und des Gewichts des Fötus, eine große Menge Fruchtwasser und sichtbare Entwicklungsanomalien (Mikrozephalie, Knochenverformung) angezeigt werden. Diese Störungen weisen mit hoher Wahrscheinlichkeit auf schwere genetische Erkrankungen hin, das Edwards-Syndrom kann jedoch nicht definitiv bestätigt werden.
- Amniozentese. Amniozentese ist eine zytologische (zelluläre) Analyse des Fruchtwassers. Der Arzt führt unter der Kontrolle eines Ultraschallgeräts vorsichtig eine spezielle Nadel ein. Die Punktion erfolgt an einer Stelle, an der sich keine Nabelschnurschlingen befinden. Mit einer Spritze wird die für die Untersuchung benötigte Menge Fruchtwasser entnommen. Der Eingriff kann in allen Schwangerschaftstrimestern durchgeführt werden, der optimale Zeitraum zur Diagnose von Chromosomenstörungen ist jedoch der Zeitraum nach der 15. Schwangerschaftswoche. Die Komplikationshäufigkeit (bis hin zum spontanen Schwangerschaftsabbruch) beträgt bis zu 1 %, daher sollte der Eingriff bei fehlender Indikation nicht durchgeführt werden. Nach dem Sammeln von Fruchtwasser wird das resultierende Material verarbeitet. Diese Flüssigkeiten enthalten Zellen von der Hautoberfläche des Babys, die Proben seiner DNA enthalten. Sie sind diejenigen, die auf das Vorliegen genetischer Krankheiten getestet werden.
- Cordozentese. Die Cordozentese ist die aussagekräftigste Methode der Pränataldiagnostik. Nach der Narkose und unter Kontrolle eines Ultraschallgeräts durchsticht der Arzt mit einer speziellen Nadel das durch die Nabelschnur verlaufende Gefäß. Auf diese Weise wird eine Blutprobe (bis zu 5 ml) des sich entwickelnden Kindes gewonnen. Die Technik zur Durchführung der Analyse ähnelt der für Erwachsene. Dieses Material kann mit hoher Genauigkeit auf verschiedene genetische Anomalien untersucht werden. Dazu gehört auch die fetale Karyotypisierung. Liegt ein zusätzliches 18. Chromosom vor, kann man von einem gesicherten Edwards-Syndrom sprechen. Es wird empfohlen, diese Analyse nach der 18. Schwangerschaftswoche (optimalerweise 22–25 Wochen) durchzuführen. Die Häufigkeit möglicher Komplikationen nach einer Cordozentese beträgt 1,5 – 2 %.
- Chorionzottenbiopsie. Das Chorion ist eine der embryonalen Membranen, die Zellen mit der genetischen Information des Fötus enthält. Bei dieser Studie wird die Gebärmutter unter Narkose durch die vordere Bauchdecke punktiert. Mit einer speziellen Biopsiezange wird eine Gewebeprobe zur Analyse entnommen. Anschließend wird eine standardmäßige genetische Untersuchung des gewonnenen Materials durchgeführt. Zur Diagnose des Edwards-Syndroms wird eine Karyotypisierung durchgeführt. Als optimaler Zeitraum für die Durchführung einer Chorionzottenbiopsie gilt die 9.–12. Schwangerschaftswoche. Die Komplikationsrate beträgt 2 – 3 %. Der Hauptvorteil, der sie von anderen Methoden unterscheidet, ist die Geschwindigkeit, mit der Ergebnisse erzielt werden (nach 2–4 Tagen).
Diagnose nach der Geburt Die Diagnose des Edwards-Syndroms nach der Geburt ist am einfachsten, schnellsten und genauesten. Leider wurde zu diesem Zeitpunkt bereits ein Kind mit einer schweren genetischen Pathologie geboren, für die es in unserer Zeit keine wirksame Behandlung gibt. Wurde die Erkrankung im Stadium der Pränataldiagnostik nicht erkannt (oder wurden keine entsprechenden Untersuchungen durchgeführt), besteht unmittelbar nach der Geburt der Verdacht auf ein Edwards-Syndrom. Das Baby ist in der Regel ausgetragen oder sogar nach der Geburt geboren, aber sein Gewicht liegt immer noch unter dem Durchschnitt. Darüber hinaus sind einige der oben genannten Geburtsfehler bemerkenswert. Wenn sie bemerkt werden, wird ein Gentest durchgeführt, um die Diagnose zu bestätigen. Dem Kind wird Blut zur Analyse entnommen. Allerdings ist die Bestätigung des Vorliegens eines Edwards-Syndroms in diesem Stadium nicht das Hauptproblem.
Die Hauptaufgabe bei der Geburt eines Kindes mit dieser Pathologie besteht darin, Anomalien in der Entwicklung der inneren Organe zu erkennen, die in der Regel in den ersten Lebensmonaten zum Tod führen. Die meisten diagnostischen Verfahren unmittelbar nach der Geburt zielen darauf ab, danach zu suchen.
Um Defekte in der Entwicklung innerer Organe zu erkennen, werden folgende Forschungsmethoden eingesetzt:
- Ultraschalluntersuchung der Bauchhöhle;
- Echokardiographie;
- allgemeiner Bluttest und biochemischer Bluttest;
- allgemeine Urinanalyse;
- Computertomographie oder Magnetresonanztomographie;
- Radiographie.
Für eine Röntgenuntersuchung gilt in der Regel das Säuglingsalter als Kontraindikation, in diesem Fall kann die Gefahr jedoch vernachlässigt werden. Tatsache ist, dass es im Moment darum geht, lebensgefährliche Pathologien sofort zu erkennen. Daher müssen Ärzte dringend alle notwendigen Informationen über bestehende Fehlbildungen einholen. Dies wird Ihnen bei der Auswahl der Behandlungstaktiken helfen. In den meisten Fällen tragen eine kompetente Patientenbetreuung und eine angemessene Pflege dazu bei, das Leben des Kindes zu verlängern.
Daraus können wir schließen, dass es viele Methoden zur Diagnose des Edwards-Syndroms gibt. Einige von ihnen (
Amniozentese, Cordozentese usw.
) bergen ein gewisses Komplikationsrisiko und werden nicht ohne besondere Indikation durchgeführt. Die Hauptindikationen sind das Vorliegen von Fällen von Chromosomenerkrankungen in der Familie und das Alter der Mutter über 35 Jahre. Das Diagnose- und Behandlungsprogramm für die Patientin in allen Stadien der Schwangerschaft kann vom behandelnden Arzt bei Bedarf geändert werden.
Prognose für Kinder mit Edwards-Syndrom
Aufgrund der vielfältigen Entwicklungsstörungen, die das Edwards-Syndrom mit sich bringt, ist die Prognose für Neugeborene mit dieser Diagnose fast immer ungünstig. Statistische Daten (
aus verschiedenen unabhängigen Studien
) Sie sagen, dass mehr als die Hälfte der Kinder (
) werden nicht älter als drei Monate. Weniger als zehn Prozent der Babys schaffen es, ihren ersten Geburtstag zu feiern. Kinder, die ein höheres Lebensalter erreichen, haben schwerwiegende gesundheitliche Probleme und benötigen ständige Pflege. Um das Leben zu verlängern, sind oft aufwendige chirurgische Eingriffe notwendig.
Nieren oder andere innere Organe. Die Korrektur von Geburtsfehlern und die fortlaufende fachmännische Betreuung sind im Wesentlichen die einzige Behandlung. Bei Kindern mit der klassischen Form des Edwards-Syndroms (
komplette Trisomie 18
) gibt es praktisch keine Chance auf eine normale Kindheit oder ein langes Leben.
Bei partieller Trisomie oder Mosaikform des Syndroms ist die Prognose etwas besser. Die durchschnittliche Lebenserwartung steigt auf mehrere Jahre. Dies erklärt sich dadurch, dass Entwicklungsanomalien bei milderen Formen nicht so schnell zum Tod des Kindes führen. Das Hauptproblem, nämlich die schwere geistige Behinderung, ist jedoch ausnahmslos allen Patienten gemeinsam. Mit Erreichen der Pubertät besteht keine Chance mehr auf Fortpflanzung (
Die Pubertät findet in der Regel nicht statt
), noch auf die Möglichkeit zu arbeiten (
sogar mechanisch, was keine besonderen Fähigkeiten erfordert
). Für die Betreuung von Kindern mit angeborenen Erkrankungen gibt es spezielle Zentren, in denen Patienten mit Edwards-Syndrom betreut und nach Möglichkeit in ihrer geistigen Entwicklung gefördert werden. Bei ausreichender Anstrengung von Ärzten und Eltern kann ein Kind, das länger als ein Jahr lebt, lernen, zu lächeln, auf Bewegungen zu reagieren, die Körperhaltung selbstständig beizubehalten oder zu essen (
in Abwesenheit von Defekten des Verdauungssystems
). Daher sind weiterhin Anzeichen einer Entwicklung zu beobachten.
Die hohe Kindersterblichkeitsrate bei dieser Krankheit erklärt sich aus einer Vielzahl von Fehlbildungen innerer Organe. Sie sind bei der Geburt sofort unsichtbar, sind aber bei fast allen Patienten vorhanden. In den ersten Lebensmonaten sterben Kinder meist an Herz- oder Atemstillstand.
Am häufigsten werden Entwicklungsstörungen in folgenden Organen und Systemen beobachtet:
- Bewegungsapparat (Knochen und Gelenke, einschließlich Schädel);
- das Herz-Kreislauf-System;
- zentrales Nervensystem;
- Verdauungssystem;
- Urogenitalsystem;
- andere Verstöße.
Bewegungsapparat Die Hauptdefekte in der Entwicklung des Bewegungsapparates sind eine abnormale Stellung der Finger und eine Krümmung der Füße. Am Hüftgelenk werden die Beine so zusammengeführt, dass sich die Knie fast berühren und die Füße leicht zur Seite schauen. Es ist nicht ungewöhnlich, dass Kinder mit Edwards-Syndrom ein ungewöhnlich kurzes Brustbein haben. Dies führt zu einer Verformung des gesamten Brustkorbs und zu Atemproblemen, die sich mit zunehmendem Alter verschlimmern, auch wenn die Lunge selbst nicht betroffen ist.
Defekte in der Entwicklung des Schädels sind hauptsächlich kosmetischer Natur. Defekte wie Gaumenspalten, Lippenspalten und Gaumenspalten führen jedoch zu ernsthaften Schwierigkeiten bei der Ernährung des Kindes. Oft wird das Kind vor Operationen zur Korrektur dieser Defekte auf parenterale Ernährung umgestellt (
in Form von Tropfern mit Nährlösungen
). Eine weitere Möglichkeit ist die Verwendung einer Gastrostomiesonde, einem speziellen Schlauch, durch den die Nahrung direkt in den Magen gelangt. Seine Installation erfordert einen separaten chirurgischen Eingriff.
Im Allgemeinen stellen Fehlbildungen des Bewegungsapparates keine unmittelbare Gefahr für das Leben des Kindes dar. Sie wirken sich jedoch indirekt auf sein Wachstum und seine Entwicklung aus. Die Häufigkeit solcher Veränderungen liegt bei Patienten mit Edwards-Syndrom bei etwa 98 %.
Das Herz-Kreislauf-System
Fehlbildungen des Herz-Kreislauf-Systems sind die häufigste Todesursache im frühen Kindesalter. Tatsache ist, dass solche Verstöße in fast 90 % der Fälle vorkommen. Meistens stören sie den Bluttransport durch den Körper ernsthaft und führen zu schweren Erkrankungen
Herzinsuffizienz
Die meisten Herzerkrankungen können chirurgisch korrigiert werden, aber nicht jedes Kind kann sich einer so komplexen Operation unterziehen.
Die häufigsten Anomalien des Herz-Kreislauf-Systems sind:
- Nichtverschluss des Vorhofseptums;
- Nichtverschluss des interventrikulären Septums;
- Verschmelzung der Klappensegel (oder umgekehrt deren Unterentwicklung);
- Aortenisthmusstenose (Verengung) der Aorta.
Alle diese Herzfehler führen zu schwerwiegenden Durchblutungsstörungen. Arterielles Blut fließt nicht in der erforderlichen Menge in das Gewebe, weshalb die Körperzellen abzusterben beginnen.
zentrales Nervensystem
Der charakteristischste Defekt des Zentralnervensystems ist die Unterentwicklung des Corpus callosum und des Kleinhirns. Dies ist die Ursache für eine Vielzahl von Störungen, einschließlich geistiger Behinderung, die bei 100 % der Kinder beobachtet wird. Darüber hinaus führen Störungen auf der Ebene des Gehirns und des Rückenmarks zu einem abnormalen Muskeltonus und einer Veranlagung dazu
Krämpfe
oder spastische Muskelkontraktionen.
Verdauungssystem
Die Inzidenz von Verdauungsstörungen beim Edwards-Syndrom beträgt bis zu 55 %. In den meisten Fällen stellen diese Entwicklungsanomalien eine ernsthafte Bedrohung für das Leben des Kindes dar, da sie es ihm nicht ermöglichen, Nährstoffe richtig aufzunehmen. Das Essen unter Umgehung der natürlichen Verdauungsorgane schwächt den Körper erheblich und verschlimmert den Zustand des Kindes.
Die häufigsten Fehlbildungen des Verdauungssystems sind:
- Meckel-Divertikel (blindes Fortsatz im Dünndarm);
- Atresie der Speiseröhre (Überwucherung ihres Lumens, wodurch die Nahrung nicht in den Magen gelangt);
- Gallengangsatresie (Ansammlung von Galle in der Blase).
Alle diese Pathologien erfordern eine chirurgische Korrektur. In den meisten Fällen hilft eine Operation nur geringfügig, das Leben des Kindes zu verlängern.
Urogenitalsystem
Die schwersten Defekte des Urogenitalsystems gehen mit einer eingeschränkten Nierenfunktion einher. In einigen Fällen wird eine Harnleiteratresie beobachtet. Die Niere auf einer Seite kann dupliziert oder mit angrenzendem Gewebe verschmolzen sein. Wenn die Filterung beeinträchtigt ist, sammeln sich mit der Zeit giftige Abfallprodukte im Körper an. Darüber hinaus kann es zu einer Erhöhung kommen
Blutdruck
und Störungen der Herzfunktion. Schwerwiegende Anomalien in der Nierenentwicklung stellen eine unmittelbare Lebensgefahr dar.
Andere Verstöße
Weitere mögliche Entwicklungsstörungen sind:
Hernien (Nabel-, Leistenbruch)
Kann auch gefunden werden
Bandscheibenvorfall der Wirbelsäule
Was zu neurologischen Problemen führen wird. Manchmal wird Mikrophthalmie in den Augen beobachtet (
kleine Augapfelgröße
Die Kombination dieser Entwicklungsstörungen führt zu einer hohen Kindersterblichkeit. Wenn das Edwards-Syndrom früh in der Schwangerschaft diagnostiziert wird, raten Ärzte in den meisten Fällen aus medizinischen Gründen zu einem Schwangerschaftsabbruch. Die endgültige Entscheidung trifft jedoch die Patientin selbst. Trotz der Schwere der Erkrankung und der schlechten Prognose hoffen viele lieber auf das Beste. Leider sind in naher Zukunft keine größeren Änderungen bei den Methoden zur Diagnose und Behandlung des Edwards-Syndroms zu erwarten.
Modern Diagnosemethoden ermöglichen es, eine Vielzahl angeborener Krankheiten bereits im Stadium der Geburt eines Kindes zu erkennen. Das Edwards-Syndrom ist eine der gefährlichsten Pathologien und führt zur Entwicklung einer Vielzahl von Erkrankungen. In der Medizin wird die Krankheit Trisomie 18 genannt.
Schauen wir uns das nun genauer an.
Was ist das Edwards-Syndrom?
Das Edwards-Syndrom ist eine Chromosomenanomalie, die zu einer ganzen Reihe von Störungen und Anomalien in der Entwicklung eines Kindes führt. Veränderungen finden auf Chromosom 18 statt. Patienten mit dem Syndrom haben zusätzliche Kopien davon. Diese Tatsache führt zu Komplikationen genetischer Natur.
Die Erkrankungshäufigkeit liegt bei 1 Fall pro 7000 gesunden Kindern. Die meisten Neugeborenen, die mit dieser Störung geboren werden, sterben in den ersten Lebenswochen. Nur etwa 10 % der Kinder leben ein Jahr. Die Krankheit führt zu schwerer geistiger Behinderung und angeborenen Schäden an inneren und äußeren Organen. Häufig kommt es zu Defekten des Herzens, der Nieren und des Gehirns. Kinder mit dieser Pathologie haben einen kleinen Kopf und Kiefer. Möglicherweise liegt eine Lippen- oder Gaumenspalte vor. Es gibt einen Klumpfuß.
Die Krankheit wurde erstmals 1960 beschrieben. Die Krankheitssymptome wurden von D. Edwards formuliert und beschrieben. Er konnte einen Zusammenhang zwischen einer Reihe von Anzeichen herstellen und entdeckte mehr als 130 mit der Krankheit verbundene Defekte. Die pathologischen Symptome treten sehr deutlich hervor. Moderne Therapiemethoden sind dagegen jedoch wirkungslos. Es gibt drei Arten der Krankheit. Die Liste umfasst:
- Einfache Trisomie. Die Krankheit wird in 90 % der Fälle erkannt.
- Mosaik-Trisomie. Beschädigte Chromosomen sind nur in einigen Zellen vorhanden.
- Translokation.
Laut Statistik werden Krankheiten bei Mädchen dreimal häufiger festgestellt als bei Jungen. Die Wahrscheinlichkeit, ein Neugeborenes mit Edwards-Syndrom zu bekommen, steigt mit dem Alter der Mutter. Die Wahrscheinlichkeit, ein solches Kind zu bekommen, ist am höchsten, wenn eine Frau über 30 Jahre alt ist. Nach 45 Jahren steigt das Risiko auf 0,7 %.
Die Pathologie beeinflusst die Lebenserwartung erheblich. Jungen werden normalerweise nur bis zu 3 Monate alt. Kranke Mädchen leben länger. Es wird darauf hingewiesen, dass solche Babys das Alter von zehn Monaten erreichten. Allerdings erreichen solche Kinder praktisch nicht das Erwachsenenalter. Zu den Ursachen für den Tod des Fötus zählen Lungeninfektionen oder Erstickungsgefahr, Funktionsstörungen des Herzens oder der Blutgefäße sowie ein gestörter oder verstopfter Darmtrakt. Alle oben genannten Phänomene entstehen als Folge einer Pathologie.
Wie das Edwards-Syndrom auf einem Foto aussieht
Moderne Methoden ermöglichen die Diagnose einer Pathologie vor der Geburt eines Kindes. In den meisten Fällen wird die Krankheit jedoch erst nach der Geburt des Neugeborenen entdeckt. Kinder mit Edwards-Syndrom weisen ausgeprägte Entwicklungsstörungen auf. Sie ermöglichen es, sofort die richtige Diagnose zu vermuten. Die Bestätigung erfolgt mittels spezieller Analyse.
Mit dem Edwards-Syndrom geborene Babys können anhand ihres Aussehens leicht von anderen unterschieden werden. Solche Kinder haben:
- verschmolzene Finger;
- schaukelnder Fuß
- veränderte Schädelform;
- abnormale Entwicklung der Geschlechtsorgane;
- Veränderung der Form des Unterkiefers;
- abnormale Entwicklung des Gaumens;
- dermatoglyphische Zeichen;
- Veränderung der Ohrenform;
- Beugestellung der Hände.
Typisch äußere Manifestation Es kommt zu einer Veränderung der Kopfform eines Neugeborenen. Kinder mit diesem Syndrom haben einen längeren, schmaleren Schädel. Die Anomalie wird durch spezielle Messungen bestätigt. Eine weitere Variante der Anomalie ist das Vorhandensein eines im Vergleich zum Rest des Körpers zu kleinen Kopfes. Dieses Phänomen betrifft insbesondere den Schädel, in dem sich das Gehirn befindet.
Alle Kinder mit einem ähnlichen Problem haben Pathologien in der Entwicklung der Ohren. Dieses Symptom wird in 95 % der Fälle beobachtet. Bei der Mosaikform des Edwards-Syndroms ist jedoch die Wahrscheinlichkeit des Auftretens einer Anomalie geringer. Bei solchen Kindern liegt die Ohrmuschel viel tiefer. Manchmal liegt es unter Augenhöhe. Die charakteristische Ausbuchtung des Knorpels, der die Ohrmuschel bildet, fehlt. Der Gehörgang selbst ist verengt. In 20–25 Prozent der Fälle kann es vollständig fehlen.
Bei Kindern bleibt der Prozess der Bildung des harten Gaumens während der intrauterinen Entwicklung unvollständig. Wo bei gesunden Menschen eine Mittelnaht vorhanden ist, liegt bei erkrankten Patienten ein Längsriss vor.
Es gibt einen Schaukelfuß. Der Defekt besteht in einer falschen Ausrichtung von Talus, Fersenbein und Strahlbein. Die Pathologie wird als Plattfußdeformität bei Kindern klassifiziert. Der Außenfuß eines Neugeborenen zeichnet sich durch die Abduktion des Fersenhöckers nach hinten aus. In diesem Fall kann es sein, dass das Fußgewölbe vollständig fehlt.
Patienten mit dieser Pathologie zeichnen sich durch ein abnormales Verhältnis der Fingerlängen aus. Normalerweise sollte der Daumen der längste sein. Bei Neugeborenen mit Edwards-Syndrom ist die Länge geringer als die Sekunde. Mit zunehmendem Alter des Kindes wird dieses Merkmal immer deutlicher. Mögliche Verschmelzung der Finger.
Es kommt zu einer Formveränderung des Unterkiefers. Bei Patienten mit Edwards-Syndrom ist das Kinn stark zurückgezogen. Dies ist auf eine Unterentwicklung des Unterkiefers zurückzuführen.
Die ersten Anzeichen des Edwards-Syndroms
Die ersten Anzeichen der Entwicklung einer Pathologie machen sich bereits im Verlauf der intrauterinen Entwicklung bemerkbar. Während der Schwangerschaft wird eine kleine Plazenta sowie Polyhydramnion beobachtet. Die Mobilität des Fötus ist eingeschränkt. Es ist nur eine einzige Nabelarterie vorhanden. Das Kind kommt mit einem Gewicht von knapp über 2 kg zur Welt. Darüber hinaus kann eine pränatale Mangelernährung vorliegen. In einigen Fällen werden auch andere Anzeichen des Edwards-Syndroms festgestellt. Daher kann es durch eine Fixierung bei der Geburt erkannt werden.
Phänotypische Merkmale sind deutlich erkennbar. Kinder haben eine niedrige Stirn und einen kleinen Mund. Es kommt zu einer Verformung der Ohren und einem hervorstehenden Nacken. Es gibt eine Gaumenspalte und eine Oberlippenspalte. Es werden Ptosis und Skelettverformungen beobachtet. Es gibt eine charakteristische Schädelform. Manchmal kommt es zu Rippenanomalien und einer angeborenen Hüftluxation. An den Ohren kann ein Tragus vorhanden sein. Der Hals des Kindes ist kurz. Es werden Strabismus und Klumpfuß beobachtet. Die Knospen sind klein. Gekreuzte Daumen werden beobachtet. Der Fuß hat die Form einer Wippe. Es gibt Papillome und Papillome auf der Haut.
Symptome des Edwards-Syndroms
Die Hauptsymptome der Krankheit sind äußere und innere Veränderungen. Sie entstehen als Folge der Entwicklung der Pathologie. Solche Kinder haben eine Gaumenspalte und eine Lippenspalte. Sie haben ein charakteristisches Aussehen und ein niedriges Geburtsgewicht. Darüber hinaus können neuropsychische Auffälligkeiten vorliegen. Die Liste der Pathologien im Zusammenhang mit der Funktion innerer Organe und motorischen Fähigkeiten umfasst:
- Ein Problem im Zusammenhang mit der Funktion des Zentralnervensystems. Bei vielen Kindern kommt es zu einer verzögerten neuropsychischen Entwicklung. Es liegt eine geistige Behinderung vor. Es besteht eine Unterentwicklung des Kleinhirns und des Corpus callosum. Es kommt zu einer Glättung oder völligen Atrophie der Gehirnwindungen.
- Probleme mit dem Herz-Kreislauf-System. Die Liste der Pathologien ist recht umfangreich. Daher können ein offenes Foramen ovale, ein ähnlicher Ductus arteriosus und ein Ventrikelseptumdefekt vorliegen.
- Pathologien der Entwicklung des Urogenitalsystems. Bei Jungen wird Kryptorchismus beobachtet. Bei Mädchen kommt es zu einer Unterentwicklung der Eierstöcke und einer Hypertrophie der Klitoris. Unabhängig vom Geschlecht kommt es zu pathologischen Veränderungen der Nieren. Es wird eine Duplikation der Harnleiter beobachtet.
- Erkrankungen des Verdauungssystems. Es wird eine gastroösophageale Refluxkrankheit beobachtet. Schluck- und Saugreflexe sind beeinträchtigt. Es kann zu einer Atresie der Speiseröhre oder des Anus kommen. Es liegt eine Verletzung der Darmlage vor.
Darüber hinaus können Schielen und Skoliose vorliegen. Es wird Muskelatrophie beobachtet.
Ursachen und Prävention des Edwards-Syndroms
Das Edwards-Syndrom tritt aufgrund einer kurzsichtigen Chromosomen-Nichtdisjunktion auf. Der Prozess findet im Stadium der Spermatogenese oder Gametogenese statt. In fast 90 % der Fälle wird die Krankheit durch ein einfaches Trypanosom repräsentiert. Seltener treten unausgeglichene Chromosomenumlagerungen oder ein Mosaiktyp der Erkrankung auf. Die Ursachen des Edwards-Syndroms sind in der Regel auf das Auftreten eines zusätzlichen mütterlichen Chromosoms im genetischen Code zurückzuführen.
Warum es zu einer solchen Chromosomenmutation kommt, konnte die Genetik bisher nicht klären. Es wird angenommen, dass dieses Phänomen zufällig ist und keine externen Faktoren Einfluss darauf haben. Es gibt jedoch immer noch eine Reihe von Gründen, die dazu beitragen Chromosomenanomalien, heute enthüllt. Die Liste umfasst:
- Genitalinfektionen bei Eltern;
- Vererbung;
- das Alter der Eltern beträgt über 45 Jahre;
- während der Schwangerschaft waren Mutter und Fötus radioaktiver Strahlung ausgesetzt;
- eine Frau nimmt seit langem Medikamente ein, die das Immun-, Hormon- und Fortpflanzungssystem beeinträchtigen (eine ähnliche Regel gilt für Männer);
- Es besteht ein langfristiger Konsum von Tabak, Drogen und Alkohol.
Wie alle genetischen Pathologien kann auch das Edwards-Syndrom nicht verhindert werden präventive Methoden. Um das Krankheitsrisiko zu verringern, können Eltern zunächst nur diagnostische Tests durchführen, um die Wahrscheinlichkeit des Auftretens einer Mutation im Voraus zu ermitteln. Darüber hinaus müssen Frau und Fötus während der Schwangerschaft engmaschig überwacht werden. Das Verfahren des vorgeburtlichen Screenings und eine Reihe anderer Studien können nicht vernachlässigt werden.
Behandlung des Edwards-Syndroms
Es muss berücksichtigt werden, dass nur jedes zehnte Kind, das mit einer ähnlichen Pathologie geboren wird, ein Jahr überlebt. Daher besteht die erste Behandlung darin, Krankheiten zu beseitigen, die das Sterberisiko erhöhen. Bei Vorliegen einer Darm- oder Analatresie kommen Maßnahmen zur Stuhlentleerung zum Einsatz. Das Kind wird über eine Sonde ernährt. Werden im Körper des Neugeborenen aktive entzündliche oder infektiöse Prozesse beobachtet, erfolgt eine therapeutische Behandlung.
Wenn das Kind noch gerettet werden kann, kann später eine Operation durchgeführt werden. Der erste Schritt besteht darin, die Gaumenspalte zu entfernen. Darüber hinaus erfolgt die Bekämpfung von Herzfehlern. Nabelschnur und werden mit chirurgischen Methoden entfernt.
Auch eine medikamentöse Therapie wird durchgeführt. Dem Kind werden Medikamente verschrieben, die es von Verstopfung, Übersäuerung und Blähungen befreien können. Eltern sollten verstehen, dass das Edwards-Syndrom zu den folgenden Pathologien beiträgt:
- Neoplasma in den Nieren;
- pulmonale Hypertonie;
- Lungenentzündung;
- Bluthochdruck;
- Sinusitis und Stirnhöhlenentzündung;
- Infektionskrankheiten des Urogenitalsystems.
Aufgrund der Vielzahl pathologischer Veränderungen in den inneren und äußeren Organen des Patienten ist die weitere Prognose ungünstig. Diejenigen Kinder, die es schaffen, bis zu einem Jahr zu leben und ein relativ erwachsenes Alter zu erreichen, werden eine offensichtliche geistige Entwicklungshemmung aufweisen. Sie werden nicht in der Lage sein, für sich selbst zu sorgen und ihre Bedürfnisse zu befriedigen natürliche Anforderungen. Diese Patienten benötigen ein Leben lang ständige Pflege und Überwachung.
Allerdings verstehen Kinder mit Behinderungen, wenn man freundlich mit ihnen spricht, mit ihnen spielt und sie tröstet. Kinder mit diesem Syndrom können lernen, selbstständig zu essen und zu lächeln. Es ist möglich, nach und nach weitere nützliche Haushaltskompetenzen zu erlernen.
Eine hohe Anzahl falsch entwickelter Pathologien innerer Organe führt zu einem hohen Prozentsatz der Kindersterblichkeit. Besteht bei einer Frau in den ersten Monaten der Schwangerschaft ein Risiko für diese Erkrankung, empfehlen Ärzte aus medizinischen Gründen einen Schwangerschaftsabbruch. Die endgültige Entscheidung liegt jedoch bei der Frau.
Nach dem Down-Syndrom ist das Edwards-Syndrom die häufigste Chromosomenstörung. Da die Prognose für Kinder, die an dieser Pathologie leiden, am enttäuschendsten ist (Tod oder tiefe geistige Behinderung für den Rest ihres Lebens), müssen Eltern unbedingt wissen, um welche Art von Abweichung es sich handelt und wann sie diagnostiziert werden kann, um die Diagnose stellen zu können rechtzeitig die richtige Entscheidung zu treffen, ob ein solches Baby im Mutterleib belassen oder die Schwangerschaft abgebrochen werden soll.
Das Trisomie-18-Syndrom oder Edwards-Syndrom ist eine Chromosomenerkrankung, die durch einen ganzen Komplex multipler, sehr schwerwiegender Entwicklungsstörungen gekennzeichnet ist. Der Grund ist die Bildung eines zusätzlichen 18. Chromosoms. Somit besteht der Karyotyp eines Patienten mit Edwards-Syndrom aus drei 18. Chromosomen statt der normalen zwei. In 90 % der Fälle handelt es sich um eine einfache Trisomie, seltener um ein Mosaik oder eine Translokation.
Die Formel, nach der der Karyotyp des Edwards-Syndroms gelesen wird, lautet wie folgt: 47,XX, 18+ (das ist ein Mädchen) und 47,XY, 18+ (das ist ein Junge).
Durch die Seiten der Geschichte. Das Edwards-Syndrom wurde erstmals 1960 von John Edwards beschrieben.
Ursachen

Die Hauptursache der Erkrankung ist das verdreifachte (und nicht wie üblich verdoppelte) Chromosom 18 im Karyotyp der Zygote. Was genau diese Pathologie hervorruft, ist den Genetikern jedoch noch unbekannt. Darüber hinaus entsteht bereits vor der Befruchtung ein zusätzliches Chromosom. Unter Ärzten herrscht die Meinung vor, dass keine anderen Tatsachen als der bloße Zufall dafür verantwortlich sind. Andererseits gibt es Risikofaktoren, die eine Eingrenzung der angeblichen Ursachen des Edwards-Syndroms in eine besondere Gruppe ermöglichen, die Paare mit Kinderwunsch im Auge behalten müssen.
- Das Alter der Mutter beträgt mehr als 45 Jahre. In diesem Fall beträgt das Risiko eines Edwards-Syndroms 0,7 %.
- Vererbung: Wenn in der Familie bereits Kinder mit ähnlichen Anomalien geboren wurden, besteht ein sehr hohes Risiko, ein Kind mit einer Pathologie des Karyotyps zur Welt zu bringen.
- Auch hier bleiben schlechte Gewohnheiten bestehen. Einige Wissenschaftler beweisen, dass die Mosaikform des Edwards-Syndroms (es ist die mildeste) durch langfristigen Drogenkonsum, eine große Menge Nikotin oder Alkohol im Körper eines Elternteils hervorgerufen werden kann.
- Langfristige Einnahme wirksamer Medikamente.
- Infektionen des Genitaltrakts.
- Strahlenbelastung führt sehr oft zu...
Dies sind nur die von Genetikern vorgeschlagenen Gründe für das Auftreten von drei statt zwei 18. Chromosomen. Sie sollten nicht als unerschütterliches Postulat verstanden werden. Risikofaktoren für das Edwards-Syndrom sind demnach das Alter der Mutter, bereits in der Familie bestehende Chromosomenerkrankungen und schlechte Angewohnheiten.
Im Vergleich dazu kommt diese Pathologie eher selten vor. Viele Eltern fragen sich, wie oft das Edwards-Syndrom diagnostiziert wird: Laut Statistik wird von 7.000 gesunden Babys 1 krankes Kind geboren. Da die Prognose für solche Kinder völlig enttäuschend ist, ist es besser, sich vorab über die Diagnose zu informieren.
Interessante Tatsache. Es ist immer noch unerklärlich, aber es werden dreimal mehr Mädchen mit dem Edwards-Syndrom geboren als Jungen.
Symptome

Um während der Schwangerschaft die richtige Entscheidung zu treffen, ob sie ihr Kind mit dieser Krankheit belassen oder nicht, müssen Eltern genau verstehen, wie Kinder mit Edwards-Syndrom bei der Geburt sind. Das klinische Bild der Pathologie ist düster und schwierig für das Herz von Mutter und Vater. Die Hauptsymptome sind bereits ab der Geburt des Babys erkennbar.
Bei der Geburt:
- geringes Gewicht (ca. 2 kg) und vorgeburtliche Unterernährung während der normalen und sogar längeren Schwangerschaftsdauer;
- Erstickung.
Verformungen von Gesicht und Schädel:
- Anomalien des Schädels, der eine dolichozephale Form, eine niedrige Stirn und einen hervorstehenden Hinterkopf hat;
- sehr kleine Mundöffnung und Unterkiefer;
- Gaumen- und Oberlippenspalte;
- schmale und kurze Lidspalten;
- Strabismus;
- Verformung der Ohren, ihre niedrige Lage relativ zum Gesicht, Dehnung in der horizontalen Ebene;
- Fehlen von Lappen und Tragus;
- verengter äußerer Gehörgang, in manchen Fällen fehlt er vollständig;
- Ptosis – schlaffe Haut;
- kurzer Hals mit charakteristischer Kragenhautfalte.
Skelettdeformitäten:
- kurzes Brustbein, wodurch die Interkostalräume kleiner werden und die Brust kürzer und breiter als normal ist;
- in 80 % der Fälle - abnormale Entwicklung der Füße: Die Fersen ragen stark hervor, das Fußgewölbe hängt durch (Schaukelfüße), die großen Zehen sind verdickt und verkürzt;
- gekreuzte Finger;
- angeborene Hüftluxation;
- Klumpfuß.
Pathologien innerer Organe:
- Herz- und Gefäßfehler;
- Kleinhirnhypoplasie;
- Magen-Darm-Erkrankungen;
- Probleme mit dem Urogenitalsystem;
- Funktionsstörungen des Zentralnervensystems;
- ausgeprägte geistige Behinderung.
Allgemeine Entwicklung:
- Schwierigkeiten beim Schlucken, Saugen und Atmen.
Ein Kind mit Edwards-Syndrom wird nie ein erfülltes Leben wie alle anderen führen können. In seinem kurzen Leben wird er viele Hindernisse überwinden und sich seiner Krankheit überhaupt nicht bewusst sein. Das Schwierigste ist für Eltern, die täglich die Entwicklung von Pathologien und Defekten beobachten müssen, mit denen ihr Baby geboren wurde. Wann wird diese Krankheit diagnostiziert?
Wow! Der Genetiker Edwards, der diese Krankheit beschrieb, identifizierte über 130 symptomatische Defekte, die für dieses Syndrom charakteristisch sind.
Diagnose

Die Diagnose des Edwards-Syndroms ist sehr wichtig, da diese Chromosomenpathologie eine medizinische Indikation für einen Schwangerschaftsabbruch darstellt. Die folgenden Methoden werden zur Identifizierung der Krankheit verwendet.
Ultraschall und Dopplerographie
Es gibt bestimmte Anzeichen des Edwards-Syndroms im Ultraschall, die Ihr Arzt beachten sollte:
- multiple fetale Entwicklungsanomalien;
- Agenesie der Nabelarterie;
- kleine Plazenta;
All dies sind jedoch indirekte Anzeichen des Edwards-Syndroms während der Schwangerschaft, die durch Daten aus anderen Studien bestätigt werden müssen.
Standardmäßiges pränatales Screening
Beinhaltet eine Blutuntersuchung auf das Vorhandensein von Serummarkern:
- von 11 bis 13 Wochen: βhCG und PAPP;
- 20 bis 24 Wochen: βhCG, freies Östriol, α-Fetoprotein.
Fällt eine Schwangere aufgrund dieser Tests in eine Hochrisikogruppe, wird ihr eine zusätzliche Diagnostik angeboten.
Invasive Diagnosemethoden

Sie werden in Ausnahmefällen durchgeführt, wenn das Risiko, ein Kind mit Edwards-Syndrom zu bekommen, sehr hoch ist. Zur invasiven Diagnostik gehören:
- Chorionzottenbiopsie;
- anschließende fetale Karyotypisierung.
Wenn die Krankheit nicht rechtzeitig erkannt wurde und ein Kind mit Edwards-Syndrom noch geboren wird, wird es sofort einer umfassenden Untersuchung unterzogen.
Diagnose nach der Geburt
Die Untersuchung eines kranken Kindes zielt darauf ab, Entwicklungsstörungen festzustellen. Ein Neugeborenes mit Edwards-Syndrom wird von verschiedenen Spezialisten untersucht:
- Neonatologe;
- Kardiologe;
- Neurologe;
- der Chirurg;
- Orthopäde;
- Urologe usw.
Die wichtigsten diagnostischen Tests, die bei einem Kind mit Edwards-Syndrom unmittelbar nach der Geburt durchgeführt werden, sind:
- Echokardiographie;
- Ultraschall der Nieren;
- Ultraschall der Bauchorgane.
Der Schwangerschaftsverlauf bei der Geburt eines Kindes mit Edwards-Syndrom unterscheidet sich in keiner Weise. Er beginnt sich auch rechtzeitig zu bewegen und ist sogar sehr aktiv; eine zusätzliche Toxikose ist nicht zu beobachten. Daher können in diesem Zeitraum nur moderne Diagnosegeräte die Krankheit erkennen. Wenn das Baby noch geboren wird, wird sein gesamtes kurzes Leben mit der ständigen Behandlung der damit verbundenen Defekte verbracht.
Meinung der Ärzte. Es werden immer mehr Mädchen mit dem Edwards-Syndrom geboren, nicht weil sie am häufigsten für diese Pathologie auf Chromosom 18 anfällig sind. Der Grund ist ihr Überleben. Es wird angenommen, dass die meisten Schwangerschaften bei Jungen mit diesem Syndrom mit einem Spontanabort oder dem intrauterinen Tod des Fötus enden.
Behandlung
Bei solchen Kindern sind Entwicklungsanomalien und alle Arten von Defekten nicht mit dem Leben vereinbar, sodass die Behandlung des Edwards-Syndroms auf eine symptomatische Behandlung beschränkt ist. Ziel ist es, grundlegende physiologische Funktionen aufrechtzuerhalten, die Lebensqualität zu verbessern und so weit wie möglich zu verlängern. Die chirurgische Korrektur angeborener Pathologien ist nach Ansicht der meisten Ärzte ungerechtfertigt und sehr riskant. In den ersten Lebenstagen beschränkt sich die Behandlung auf folgende Aktivitäten:
- Sondenernährung aufgrund von Schluck- und Saugbeschwerden;
- langfristige mechanische Beatmung aufgrund von Atemproblemen.
Nach der Entlassung sollten Eltern dem kranken Baby (wenn möglich) Folgendes zur Verfügung stellen:
- organisierte, fast professionelle Betreuung: Es ist gut, wenn neben dem Kind eine Person (Nanny) mit medizinischer Ausbildung steht;
- gute Ernährung;
- Regelmäßige Überwachung durch verschiedene Kinderärzte.
Wir müssen sofort bedenken, dass die Betreuung eines Kindes mit Edwards-Syndrom viel Geduld und Kraft erfordert.
Prognosen

Eltern, deren Kind Träger des Edwards-Syndroms ist, sollten wissen, welche Prognose sie erwartet. Wie oben erwähnt sind sie sehr enttäuschend:
- die Lebenserwartung solcher Kinder ist sehr kurz;
- 60 % sterben vor dem dritten Lebensmonat (Jungen fallen am häufigsten in diese Gruppe);
- weitere 10 % überleben bis zu einem Jahr;
- Nur 1 % der Geborenen erreichen das 10. Lebensjahr;
- die häufigste Todesursache ist eine Herzfunktionsstörung oder ein Atemstillstand;
- alle Kinder mit Edwards-Syndrom bleiben bis zum Ende ihrer Tage stark oligofren;
- der Körper solcher Kinder ist sehr geschwächt und anfällig für alle möglichen Krankheiten, weshalb sie häufig an Harnwegsinfektionen, Bindehautentzündung, mittlerer Lungenentzündung und Lungenentzündung leiden;
- Die verbalen Kommunikationsfähigkeiten solcher Kinder sind sehr begrenzt;
- sie können auf die Worte ihrer Eltern reagieren;
- manche lächeln sogar, erkennen Betreuer und interagieren mit ihnen;
- in der Lage, Fähigkeiten wie Selbsternährung und Kopfheben zu erwerben.
Wenn während der Schwangerschaft bei dem Kind das Edwards-Syndrom diagnostiziert wurde, zwingt dies die Eltern zu einer verantwortungsvollen Entscheidung (den Zeitpunkt der Diagnose verschiedener fetaler Pathologien finden Sie in). Werden sie in der Lage sein, ein schwerkrankes Kind zu betreuen, das an mehreren Pathologien leidet? Oder ist es sinnvoll, eine solche Schwangerschaft abzubrechen? Hier kann nur das Paar selbst durch gemeinsame Anstrengungen den einzig richtigen Ausweg für sich finden.
Im Jahr 1971 Auf der Pariser Konferenz wurde eine spezielle Nomenklatur zur Erfassung des menschlichen Karyotyps verabschiedet.
Normaler menschlicher Karyotyp:
46,XX - Frau; 46, XY - männlich.
Karyotyp für Polyploidie:
69,XXX; 69,ХХУ - Triploidie;
92,XXXX; 92,ХХХУ - Tetraploidie.
Karitätstyp für Monosomie:
45,XO ist die einzige Monosomie, die bei lebenden Menschen möglich ist (Schereshevsky-Turner-Syndrom).
Karyotyp für autosomale Trisomie:
47,XX,+21 oder 47,XY,+21 – Trisomie auf Chromosom 21 (Down-Syndrom);
47,XX,+13 oder 47,XY.+13 - Trisomie auf Chromosom 13 (Patau-Syndrom);
47,XX.+18 oder 47,XY,+18 – Trisomie auf Chromosom 18 (Ewards-Syndrom).
Karyotyp für Trisomie der Geschlechtschromosomen:
47.XXX - Trisomie X bei einer Frau;
47, XYU – Trisomie U bei einem Mann.
47,ХХУ – Klinefelter-Syndrom.
Tetrasomie und Pentasomie der Geschlechtschromosomen:
48,ХХХХ – Tetrasomie X;
49,ХХХХХ - Pentasomie X;
48,ХХХУ; 49,ХХХХУ – Varianten des Klinefelter-Syndroms;
48,HUUU; 49,HUUUU – Varianten des Polysomie-U-Syndroms bei Männern.
Karyotyp für Chromosomenaberrationen:
46,ХХ,del 5p - - Deletion des kurzen Arms von Chromosom 5 (Crying-Cat-Syndrom) bei einer Frau;
46,XY,del 4p - - Deletion des kurzen Arms von Chromosom 4 (Wolf-Hirschhorn-Syndrom) bei einem Mann;
46,Х,i (Xq) – Isochromosom X entlang des langen Arms einer Frau;
46,XY,r (18) – radiales Chromosom 18 bei Männern;
45,ХХ, -Ä,-У,+ t (Äq, Уq) – eine ausgewogene Robertsonsche Translokation, die durch die Verbindung der langen Arme eines D- und eines Y-Chromosoms bei einer Frau entsteht.
Karyotyp für Mosaik:
45,Х/46,ХХ oder 45,Х/46,ХХ – einige Zellen haben einen normalen Karyotyp (46,ХХ) und einige mit Monosomie X (45,ХХ). Wir sprechen von der Mosaikform des Shereshevsky-Turner-Syndroms;
47,ХХ,+21/ 46,ХХ – Mosaikform des Down-Syndroms.
Pathogenese chromosomaler Erkrankungen.
Bei Chromosomenerkrankungen liegt in der Regel ein Ungleichgewicht in einer Vielzahl von Genen vor. Der veränderte Genotyp manifestiert sich in der embryonalen Entwicklungsphase. Die frühesten Stadien der Zygotenspaltung werden durch im Ei angesammelte Substanzen gesteuert. Dann werden die eigenen Gene der Zygote aktiviert. Insgesamt wirken in der Embryonalperiode etwa 1000 Gene, die für verschiedene Stadien der Ontogenese verantwortlich sind. Sie sind über alle Chromosomen verteilt. Bei genomischen und chromosomalen Mutationen wird das Gleichgewicht einer Vielzahl von Genen gestört, darunter auch Gene, die die Embryonalentwicklung regulieren. Dies führt zwangsläufig zu einer Störung der Histogenese und Organogenese. Es entstehen Entwicklungsstörungen. Häufiger erweisen sich Verstöße als lebensunvereinbar, was zum intrauterinen Tod des Embryos führt. Es kommt seltener vor, dass ein Kind mit Entwicklungsstörungen geboren wird.
35 bis 50 % (jetzt schreiben sie bis zu 70 %) der menschlichen Embryonen sterben im Blastozystenstadium, d. h. vor der Implantation. Ein großer Prozentsatz von ihnen weist Chromosomenumlagerungen auf. Nach der Implantation beträgt der Gesamtbeitrag von Chromosomenanomalien zum intrauterinen Tod beim Menschen 45 %. Je früher die Schwangerschaft abgebrochen wird, desto wahrscheinlicher ist es, dass sie auf ein chromosomales Ungleichgewicht zurückzuführen ist.
Kommt es in den ersten 2–4 Wochen zu einer Abtreibung, wird bei 60–70 % der Abtreibungen ein chromosomales Ungleichgewicht beobachtet. Im ersten Trimester – bei 50 %, im 2. Trimester – bei 30 %, in der 20. bis 27. Woche – bei 7 % und schließlich bei 6 % der Totgeburten werden durch eine Chromosomenpathologie verursacht.
Sind Störungen der Embryonalentwicklung mit dem Leben vereinbar, kommt ein Kind mit Entwicklungsstörungen zur Welt.
U 1 % lebendig Bei Neugeborenen werden bestimmte Chromosomenerkrankungen diagnostiziert.
Klinisch äußern sich Chromosomenerkrankungen als Syndrome multipler angeborener Fehlbildungen. Fast alle von ihnen sind zum Zeitpunkt der Geburt ausgebildet. Ausnahmen sind Störungen bei der Ausbildung der Geschlechtsmerkmale aufgrund eines Ungleichgewichts der Geschlechtschromosomen. Einige ihrer Symptome treten im Jugendalter auf. Genetiker vergleichen Chromosomenerkrankungen mit der Asche nach einem Brand. Feuer entsteht während der Embryonalperiode. Zum Zeitpunkt der Geburt wird der endgültige Phänotyp gebildet (Feuerbrände). Es lässt sich nichts mehr reparieren. Eine kosmetische Korrektur und Operation können Sie nur bei einem Patienten mit einer Entwicklungsstörung durchführen (sofern das Syndrom mit dem Leben vereinbar ist).
Da bei CB die frühen Stadien der Embryonalentwicklung gestört sind, sind viele Organe und Organsysteme gleichzeitig betroffen. Dadurch ähnelt sich das Krankheitsbild vieler Chromosomenerkrankungen. Je größer das Chromosomenungleichgewicht ist, desto unspezifischer ist das Bild.
Jede Chromosomenerkrankung ist durch Polymorphismus gekennzeichnet, weil Der individuelle Genotyp von Individuen beeinflusst die Genexpression.
KLINISCH-ZYTOGENETISCHE EIGENSCHAFTEN DER HÄUFIGSTEN CHROMOSOMLEN ERKRANKUNGEN