Trisomie régulière sur le chromosome 18. Qu'est-ce que le diagnostic du syndrome d'Edwards ?
Lire aussi
La plupart des femmes savent bien ce qu'est le syndrome de Down. Et pendant la grossesse, beaucoup apprennent cela très rarement, mais il existe un autre trouble chromosomique appelé syndrome d'Edwards. Et beaucoup se demandent comment savoir à quel point le risque d'avoir un enfant atteint du syndrome d'Edwards est élevé et comment une telle pathologie est diagnostiquée pendant la grossesse?
Qu'est-ce que le syndrome d'Edwards ?
Le syndrome d'Edwards est une maladie génétique caractérisée par une duplication (trisomie) du chromosome ХVIII et se manifeste par un certain nombre de malformations caractéristiques du fœtus pendant la grossesse, entraînant souvent la mort de l'enfant ou son handicap. C'est-à-dire que chez un enfant, au lieu de 46 chromosomes, 47 sont formés, ce chromosome supplémentaire donne un autre nom à la maladie - la trisomie 18. Le syndrome a été nommé d'après le chercheur John Edwards, qui l'a décrit pour la première fois en 1960.
Pourquoi le syndrome d'Edwards se produit - causes de la pathologie
Même si les parents sont en bonne santé et qu'il n'y a pas une telle pathologie dans les antécédents familiaux, un enfant avec le chromosome 18 peut naître de n'importe quelle femme.
Comme vous le savez, dans chaque cellule humaine, il y a 46 chromosomes, et dans les cellules germinales femelles et mâles, 23 chromosomes chacune, qui, lorsqu'ils sont combinés lors de la fécondation d'un ovule, donnent également un total de 46 chromosomes. Si nous parlons du syndrome d'Edwards, les causes de son apparition sont inconnues.
Actuellement, on sait seulement qu'à la suite de certaines mutations génétiques, un 47e chromosome supplémentaire apparaît (un chromosome supplémentaire dans la 18e paire de chromosomes, qui devient ainsi non pas 2, mais 3).
Dans 95 % des cas de développement du syndrome d'Edwards, il s'agit du 18e chromosome supplémentaire (trisomie) dans les cellules, mais dans 2 % des cas, il n'y a qu'un "allongement" du 18e chromosome (translocation), lorsque le nombre total de chromosomes reste normal et égal à 46.
Dans 3% des cas de syndrome d'Edwards, une «trisomie en mosaïque» se produit, lorsqu'un 47e chromosome supplémentaire se trouve dans le corps, non pas dans toutes les cellules, mais seulement dans une certaine partie de celui-ci. Cliniquement, les 3 variantes du syndrome d'Edwards se déroulent presque de la même manière, cependant, la première variante peut différer dans une évolution plus sévère de la maladie.
Quelle est la fréquence de cette pathologie ?
Les enfants atteints du syndrome d'Edwards meurent in utero dans environ 60 % des cas. Malgré cela, parmi les maladies génétiques, ce syndrome chez les nourrissons survivants est assez courant, juste derrière le syndrome de Down en termes de fréquence d'apparition. La prévalence du syndrome d'Edwards est de 1 cas sur 3 à 8 000 enfants.
On pense que chez les filles, cette maladie survient 3 fois plus souvent et que le risque de syndrome d'Edwards augmente considérablement si une femme enceinte a 30 ans ou plus.
La mortalité dans le syndrome d'Edwards au cours de la première année de vie est d'environ 90% et l'espérance de vie moyenne dans les cas graves de la maladie chez les garçons est de 2 à 3 mois et chez les filles de 10 mois, et seuls quelques-uns survivent jusqu'à l'âge adulte. Le plus souvent, les enfants atteints du syndrome d'Edwards meurent d'étouffement, de pneumonie, d'insuffisance cardiovasculaire ou d'obstruction intestinale - complications causées par des malformations congénitales.
Comment le syndrome se manifeste-t-il chez un enfant ?
Les symptômes du syndrome d'Edwards peuvent être divisés en plusieurs grands groupes :
Le premier groupe comprend des symptômes caractéristiques de l'apparence de l'enfant:
- A la naissance, faible poids corporel (environ 2100 - 2200 grammes)
- Tête disproportionnellement petite
- Défauts de développement des mâchoires supérieure ou inférieure (micrognathie)
- Distorsion de la forme du visage et formation de malocclusion
- Fente palatine (fente palatine) ou fente labiale (fente labiale)
- Les doigts de la main sont serrés, ils sont inégaux dans le poing
- Oreilles attachées bas
- Sangle ou fusion complète des doigts des membres inférieurs
- pied bot congénital
- "Bascule du pied"
- Fissure buccale relativement petite (microstomie)
Le deuxième groupe comprend des signes de perturbation les organes internes, motricité et développement neuropsychique :
- La présence de malformations cardiaques congénitales, par exemple, un foramen ovale ouvert, une communication interventriculaire, un canal artériel ouvert, etc.
- Développement de hernies inguinales ou ombilicales.
- Organes digestifs : reflux gastro-œsophagien, altération du réflexe de déglutition et de succion, atrésie de l'œsophage ou anus, diverticule de Meckel, violation de l'emplacement de l'intestin.
- Système nerveux central : retard du développement neuropsychique, retard mental, sous-développement du cervelet, du corps calleux, lissage ou atrophie des circonvolutions cérébrales.
- Système génito-urinaire: cryptorchidie, hypospadias chez les garçons, hypertrophie clitoridienne, sous-développement des ovaires chez les filles, quel que soit le sexe - rein en fer à cheval ou segmenté, doublement des uretères.
- Strabisme, scoliose, atrophie musculaire.
Comment trouver la pathologie pendant la grossesse - diagnostic
Le syndrome de Patau, le syndrome d'Edwards et d'autres trisomies sont mieux identifiés avant la naissance du bébé. En règle générale, le diagnostic prénatal de ce syndrome est réalisé en 2 étapes:
- Pendant une période de 11 à 13 semaines (dépistage, qui repose sur divers tests biochimiques chez une femme).
- Détermination du caryotype du fœtus chez les femmes enceintes de leur groupe à risque.
A 11-13 semaines, le taux de certaines protéines sanguines est déterminé dans le sang de la femme : la β-hCG (sous-unité β de l'hormone chorionique humaine) et la protéine plasmatique A associée à la grossesse. Ensuite, en tenant compte de ces données, l'âge de la femme enceinte, le risque d'avoir un enfant atteint du syndrome d'Edwards est calculé et un groupe à risque pour les femmes enceintes est formé.
De plus, dans le groupe à risque à une date ultérieure, du matériel est prélevé sur le fœtus pour établir un diagnostic précis: à 8–12 semaines, il s'agit d'une biopsie des villosités choriales, à 14–18 - amniocentèse (étude du liquide amniotique), après 20 semaines - cordocentèse (prélèvement de sang intra-utérin du fœtus du cordon ombilical avec guidage échographique). Après cela, la présence ou l'absence d'un 18ème chromosome supplémentaire est déterminée dans le matériau obtenu à l'aide de KF-PCR (réaction en chaîne par polymérase fluorescente quantitative).
Si une femme enceinte n'a pas réussi un examen de dépistage génétique, alors pour plus dates ultérieures le diagnostic préliminaire du syndrome d'Edwards est réalisé à l'aide d'une échographie. Autres signes indirects, sur la base desquels un syndrome d'Edwards peut être suspecté ultérieurement :
- La présence d'anomalies dans le développement des os et des tissus mous de la tête ("fente palatine", microcéphalie, atterrissage bas des oreillettes, "fente labiale", etc.).
- Détection de défauts de la part du système cardiovasculaire, système génito-urinaire ainsi que le système musculo-squelettique.
Signes diagnostiques du syndrome chez un enfant
Après la naissance d'un enfant, les principaux signes diagnostiques de la présence du syndrome d'Edwards sont les suivants :

Signes d'une image dermatographique caractéristique:
- pli de flexion distal non développé sur les doigts
- la présence dans 1/3 des cas d'une rainure palmaire transversale
- arcs sur le bout des doigts
- modification du motif cutané de la paume : emplacement distal du triradius axial et augmentation du nombre de crêtes.
Syndrome d'Edwards à l'échographie - kystes du plexus vasculaire
Dans les premiers stades, il est extrêmement difficile de suspecter le syndrome d'Edwards à l'échographie, cependant, à 12 semaines de grossesse, des symptômes de nature indirecte qui le caractérisent sont déjà révélés. :
- Signes de retard de croissance fœtale
- Bradycardie (diminution du rythme cardiaque fœtal)
- Omphalocèle (présence d'une hernie de la cavité abdominale)
- Pas de visualisation des osselets nasaux
- Il y a une artère dans le cordon ombilical, pas 2
Également à l'échographie, les kystes du plexus vasculaire, qui sont des cavités contenant du liquide, peuvent être détectés. En eux-mêmes, ils ne constituent pas une menace pour la santé et disparaissent à la 26e semaine de grossesse. Cependant, de tels kystes accompagnent très souvent diverses maladies génétiques, par exemple le syndrome d'Edwards (dans ce cas, des kystes se retrouvent chez 1/3 des enfants souffrant de cette pathologie), donc, si de tels kystes sont découverts, le médecin orientera la femme enceinte femme à une consultation génétique pour examen.
Étant donné que les enfants atteints du syndrome d'Edwards vivent rarement jusqu'à un an, le traitement vise d'abord à corriger les malformations potentiellement mortelles :
- restauration du passage des aliments dans l'atrésie de l'intestin ou de l'anus
- alimentation par sonde en l'absence de réflexes de déglutition et de succion
- thérapie antibactérienne et anti-inflammatoire pour la pneumonie
Avec une évolution relativement favorable de la maladie, certaines anomalies et malformations sont corrigées : traitement chirurgical de la "fente palatine", malformations cardiaques, hernie inguinale ou ombilicale, ainsi que traitement médicamenteux symptomatique (prescription de laxatifs contre la constipation, "anti-mousse" pour les flatulences, etc.) . 
Les enfants atteints du syndrome d'Edwards sont sujets à des maladies telles que :
- otite moyenne
- cancer du rein (tumeur de Wilms)
- pneumonie
- conjonctivite
- hypertension pulmonaire
- apnée
- hypertension artérielle
- frontite, sinusite
- infections des voies urinaires
Par conséquent, le traitement des patients atteints du syndrome d'Edwards comprend un diagnostic et un traitement rapides de ces maladies.
Pronostic pour un enfant
Dans la plupart des cas, le pronostic est mauvais. Les quelques enfants atteints du syndrome d'Edwards qui survivent jusqu'à l'âge adulte ont de graves handicaps mentaux et nécessitent constamment des soins et une surveillance extérieurs. Cependant, avec des activités appropriées, ils sont capables de répondre au confort, de sourire, de manger de manière autonome et d'interagir avec les soignants, en acquérant diverses compétences et capacités.
Syndrome d'Edwards

Syndrome d'Edwards- une maladie chromosomique causée par une trisomie sur le 18ème chromosome et accompagnée de multiples malformations. Le syndrome d'Edwards se caractérise par des caractéristiques phénotypiques particulières (forme dolichocéphalique du crâne, microphtalmie, sous-développement des oreillettes, microrétrognathie, etc.), des anomalies des systèmes musculo-squelettique, cardiovasculaire, digestif, génito-urinaire et du système nerveux central. Le syndrome d'Edwards peut être diagnostiqué au stade de la grossesse (dépistage échographique, diagnostic prénatal invasif) ou après la naissance d'un enfant sur la base de signes extérieurs et d'études cytogénétiques. Les enfants atteints du syndrome d'Edwards ont besoin d'un traitement symptomatique et de bons soins.
Syndrome d'Edwards

Le syndrome d'Edwards est une aberration chromosomique quantitative dans laquelle il existe une trisomie partielle ou complète sur le 18ème autosome. Le syndrome a été nommé d'après le généticien J. Edwards, qui a décrit la maladie en détail en 1960 et identifié plus de 130 défauts symptomatiques caractéristiques de cette pathologie. Le syndrome d'Edwards est le deuxième trouble chromosomique le plus fréquent après le syndrome de Down ; le taux de natalité des enfants atteints du syndrome d'Edwards est de 1:5000-7000. Environ les trois quarts de tous les patients atteints du syndrome d'Edwards sont des filles; on suppose que la plupart des grossesses avec un fœtus de sexe masculin se terminent par une mort fœtale et un avortement spontané.
Causes du syndrome d'Edwards
Le développement du syndrome d'Edwards s'explique par des troubles chromosomiques survenant au stade de la gamétogenèse (ovogenèse ou spermatogenèse) ou du clivage zygote et entraînant une augmentation du nombre de chromosomes de la 18e paire. Dans 80 à 90 % des cas, les variants cytogénétiques du syndrome d'Edwards sont représentés par une simple trisomie 18, moins souvent par une forme mosaïque ou des remaniements déséquilibrés (translocations).
La cause de la trisomie complète est la non-disjonction méiotique des chromosomes. Dans presque tous les cas, le chromosome supplémentaire est d'origine maternelle. Cette variante du syndrome d'Edwards est la plus sévère dans ses manifestations et défavorable en termes de pronostic. L'émergence du mosaïcisme est associée à la non-disjonction des chromosomes à un stade précoce du clivage du zygote. Dans ce cas, toutes les cellules du fœtus ne contiendront pas le chromosome supplémentaire, mais seulement une partie d'entre elles. Translocation - l'attachement d'une partie du 18e chromosome à une autre paire peut se produire à la fois pendant la maturation des gamètes et après la fécondation. Dans ce cas, les cellules du corps contiennent deux 18èmes chromosomes homologues et sa partie supplémentaire attachée à un autre chromosome.
Comme pour le syndrome de Down, l'âge de la mère est le facteur de risque le plus important d'avoir un enfant atteint du syndrome d'Edwards. À Cas rares les parents peuvent être porteurs d'une translocation équilibrée.
Symptômes du syndrome d'Edwards
Pendant la grossesse, il y a des polyhydramnios, une faible activité fœtale, un petit placenta, la seule artère ombilicale. Un enfant atteint du syndrome d'Edwards naît avec un faible poids corporel (environ 2170 g) et une malnutrition prénatale pendant une grossesse à terme ou même après terme. Chez certains enfants, l'état d'asphyxie à la naissance est déterminé.
Les nouveau-nés atteints du syndrome d'Edwards présentent des caractéristiques phénotypiques caractéristiques suggérant cette pathologie chromosomique. Tout d'abord, l'attention est attirée sur la forme dolichocéphale du crâne avec une prédominance de la taille longitudinale sur transversale, front bas, occiput saillant, micrognathie, petite bouche, microphtalmie. Les enfants atteints du syndrome d'Edwards ont souvent une fente labiale et palatine, un épicanthe, un ptosis, une exophtalmie, un strabisme, un cou court avec un pli cutané excessif. Les déformations typiques du pavillon comprennent de petits lobes d'oreille, un manque de tragus, des conduits auditifs étroits et des oreilles basses.
L'apparence des enfants est complétée par des déformations squelettiques caractéristiques du syndrome d'Edwards - doigts croisés, sternum raccourci, anomalies des côtes, luxation congénitale de la hanche, pied bot, pied basculant, syndactylie des pieds, etc. De nombreux enfants ont des hémangiomes et des papillomes cutanés .
Avec le syndrome d'Edwards, il existe de multiples anomalies graves de presque tous les systèmes du corps. Les malformations cardiaques congénitales peuvent être représentées par des anomalies des septa interventriculaires et interauriculaires, une coarctation de l'aorte, une transposition des vaisseaux principaux, une dysplasie valvulaire, une tétralogie de Fallot, un drainage anormal des veines pulmonaires, une dextrocardie, etc. Avec le syndrome d'Edwards, la pathologie de le développement du tractus gastro-intestinal peut être détecté: hernie diaphragmatique, ombilicale et inguinale, diverticule de Meckel, fistules trachéo-œsophagiennes, sténose pylorique, atrésie de l'iléon et de l'anus. Les anomalies les plus courantes de l'appareil génito-urinaire chez les enfants atteints du syndrome d'Edwards sont le rein en fer à cheval, l'hydronéphrose, les diverticules vésicaux, l'hypospadias et la cryptorchidie (chez les garçons), l'utérus bicorne, le septum intra-utérin et l'hypertrophie clitoridienne (chez les filles).
Malformations de la centrale système nerveux caractérisé par la présence de microcéphalie, méningomyélocèle, hydrocéphalie, anomalies d'Arnold-Chiari, kystes du plexus arachnoïdien, hypoplasie du cervelet et du corps calleux. Tous les enfants survivants atteints du syndrome d'Edwards ont des handicaps intellectuels- oligophrénie dans le degré d'imbécillité profonde ou d'idiotie.
Les nouveau-nés atteints du syndrome d'Edwards ont des difficultés à téter, à avaler et à respirer, nécessitant une alimentation par sonde ou une ventilation mécanique à long terme. En règle générale, les enfants atteints du syndrome d'Edwards meurent au cours de la première année de vie en raison de malformations congénitales graves et de complications associées (insuffisance cardiovasculaire et respiratoire, pneumonie, occlusion intestinale, etc.).
Diagnostic du syndrome d'Edwards
La tâche diagnostique la plus importante est la détection prénatale du syndrome d'Edwards chez le fœtus, car cette pathologie est une indication médicale pour l'interruption artificielle de la grossesse. Il est possible de suspecter la présence d'un syndrome d'Edwards dans le processus d'échographie du fœtus et de dopplerographie du flux sanguin utéroplacentaire par des signes indirects (anomalies multiples du développement du fœtus, agénésie de l'artère ombilicale, petit placenta, polyhydramnios, etc. .).
Le dépistage prénatal standard a la plus grande importance diagnostique, y compris un test sanguin pour les marqueurs sériques : βhCG et PAPP à 11-13 semaines de gestation ; βhCG, alpha-foetoprotéine et estriol libre à 20-24 semaines de gestation.
Lors de l'évaluation du risque d'avoir un enfant atteint du syndrome d'Edwards, les données de dépistage biochimiques et échographiques, l'âge gestationnel, l'âge et le poids corporel de la femme sont pris en compte. Les femmes enceintes appartenant au groupe à haut risque se voient proposer un diagnostic prénatal invasif (biopsie chorionique, amniocentèse, cordocentèse) suivi d'un caryotypage fœtal.
En cas de naissance d'un enfant vivant atteint du syndrome d'Edwards, un examen complet le plus précoce possible est nécessaire, visant à identifier les malformations graves. Un nouveau-né atteint du syndrome d'Edwards doit être examiné par un néonatologiste, un cardiologue pédiatrique, un neurologue pédiatrique, un chirurgien pédiatrique, un orthopédiste pédiatrique, un urologue pédiatrique, etc. Les études diagnostiques les plus importantes à effectuer sur un enfant atteint du syndrome d'Edwards dans les premières heures de vie sont l'échocardiographie, l'échographie de la cavité abdominale et l'échographie des reins.
Traitement du syndrome d'Edwards
Les anomalies du développement étant dans la plupart des cas incompatibles avec la vie, le traitement des enfants atteints du syndrome d'Edwards se réduit à fournir des soins symptomatiques visant à maintenir les fonctions physiologiques, à prolonger la vie et à améliorer sa qualité. La correction chirurgicale des malformations congénitales est généralement risquée et injustifiée.
Étant donné que les enfants atteints du syndrome d'Edwards sont faibles et sujets à de fréquentes infections des voies urinaires, otites moyennes, conjonctivites, sinusites, pneumonies, etc., ils ont besoin de soins soigneusement organisés, d'une bonne nutrition et d'une surveillance régulière par un pédiatre.
Prévision et prévention du syndrome d'Edwards
Dans tous les cas, le pronostic du syndrome d'Edwards est extrêmement défavorable: en moyenne, les garçons vivent 2 à 3 mois, les filles - 10 mois. Jusqu'à 1 an, seuls 10% des patients survivent, jusqu'à 10 ans - pas plus de 1%. Les enfants atteints d'une forme mosaïque du syndrome d'Edwards ont des chances de survie relativement favorables.
Le risque d'avoir un enfant atteint du syndrome d'Edwards existe théoriquement dans tout couple marié ; on sait que cette probabilité est plus élevée chez les parents plus âgés (pour les femmes de plus de 45 ans - 0,7%). Afin de détecter à temps une pathologie chromosomique chez le fœtus, le dépistage prénatal, qui fait partie du programme d'introduction à la grossesse, ne doit pas être négligé.
Syndrome d'Edwards
Notre équipe de professionnels répondra à vos questions
Syndrome d'Edwards (trisomie sur le chromosome 18) est la deuxième anomalie chromosomique la plus fréquente après le syndrome de Down. La fréquence du syndrome d'Edwards est de 1:5000-1:7000 nouveau-nés. Les filles atteintes du syndrome d'Edwards naissent trois fois plus souvent que les garçons.
Le «gold standard» pour détecter les troubles chromosomiques dans le monde a longtemps été et continue d'être la méthode de caryotypage avec coloration différentielle des chromosomes. Cette méthode permet d'analyser le caryotype dans son ensemble et de déterminer de grands réarrangements chromosomiques (au moins 5 à 10 millions de paires de bases). Cependant, elle présente un certain nombre de limites, telles que l'intensité de la main-d'œuvre, la durée (1 à 2 semaines), les exigences élevées en matière de qualifications et d'expérience du spécialiste menant l'étude et, dans certains cas, des problèmes techniques (quantité et qualité insuffisantes de le matériel étudié, l'absence de mitoses ou de croissance de culture).
La méthode quantitative de réaction en chaîne par polymérase fluorescente (QF-PCR), de plus en plus utilisée pour diagnostiquer les aneuploïdies, dont le syndrome d'Edwards, est dépourvue de ces lacunes (Fig. 1). Cette méthode a une fiabilité comparable à celle du caryotypage standard, est plus rapide, moins chère, moins exigeante sur la quantité et la qualité du matériel (puisqu'elle n'est pas associée à la croissance de la culture cellulaire) et permet d'analyser simultanément un grand nombre de échantillons. Cependant, la méthode CF-PCR présente également des limites : dans les cas de mosaïcisme, elle ne peut détecter qu'un mosaïcisme de haut niveau (à partir de 20 %), de plus, elle ne peut pas exclure la présence d'anomalies chromosomiques plus rares pouvant être associées à des malformations fœtales. Lors du diagnostic prénatal du syndrome d'Edwards, en plus du matériel du fœtus, il est nécessaire de fournir le matériel biologique de la mère afin d'exclure la possibilité d'obtenir un faux résultat négatif en raison d'un échantillonnage inapproprié du matériel fœtal. L'analyse du matériel fœtal est effectuée dans les trois jours ouvrables.
Avec le syndrome d'Edwards, il y a un retard prononcé dans le développement prénatal, les enfants naissent avec une malnutrition prénatale (le poids corporel moyen à la naissance est de 2340 g). Les manifestations externes du syndrome d'Edwards sont diverses (Fig. 2). Les plus typiques sont un retard du développement psychomoteur, une hypoplasie des muscles squelettiques et du tissu adipeux sous-cutané, des malformations cardiaques congénitales, des anomalies de la structure de la face et du crâne (dolichocéphalie, microphtalmie, raccourcissement des fissures palpébrales, position basse des oreillettes, micrognathie, menton en pente), multiples déformations des mains et des pieds, anomalies du développement du tractus gastro-intestinal, de l'appareil génito-urinaire et du système nerveux central (hernie spinale, hypoplasie du corps calleux et du cervelet). L'espérance de vie des enfants est fortement réduite : 90 % d'entre eux meurent avant un an de complications dues à des malformations congénitales (asphyxie, pneumonie, occlusion intestinale, insuffisance cardiovasculaire).
La raison du développement du syndrome d'Edwards est le triplement du chromosome 18. La trisomie sur le chromosome 18 est un cas particulier d'aneuploïdie - la présence dans le génome d'un ensemble de chromosomes différent du standard pour cette espèce et non multiple de celui-ci . La trisomie 18 est généralement causée par la non-disjonction des chromosomes lors de la formation des cellules germinales des parents (ovules et spermatozoïdes), à la suite de quoi l'enfant reçoit un 18e chromosome supplémentaire de la mère ou du père. Dans ce cas, toutes les cellules du corps de l'enfant seront porteuses de l'anomalie. Dans le cas où la non-disjonction des chromosomes se produit lors de la division de n'importe quelle cellule de l'embryon, une variante en mosaïque du syndrome d'Edwards est observée (10% des cas).
Le risque d'avoir des enfants atteints du syndrome d'Edwards, selon diverses données de la littérature, ne change pas ou augmente légèrement avec l'âge de la femme enceinte.
Le diagnostic prénatal du syndrome d'Edwards comprend deux étapes. Au premier stade, à un âge gestationnel de 11-13 semaines, un dépistage est effectué, qui repose principalement sur des paramètres biochimiques, car dans les premiers stades, l'échographie ne permet pas de détecter d'anomalies grossières du développement dans le cas du syndrome d'Edwards, qui ne peut être détecté qu'à 20-24 semaines. Analyse biochimique du taux de certaines protéines dans le sang d'une femme enceinte (sous-unité β libre de l'hormone chorionique humaine (β-hCG) et protéine A plasmatique associée à la grossesse (protéine A plasmatique associée à la grossesse, PAPP-A)), compte tenu de son âge, permet de calculer pour elle le risque d'avoir un enfant malade. Cependant, ces méthodes ne permettent pas de poser un diagnostic précis et, à la suite du dépistage, un groupe à risque de femmes enceintes présentant une probabilité accrue de donner naissance à un patient atteint du syndrome d'Edwards est formé. À la deuxième étape, une procédure invasive est effectuée dans le groupe à risque pour obtenir le matériel fœtal nécessaire pour déterminer avec précision l'état du fœtus. Selon l'âge gestationnel, il peut s'agir d'une biopsie des villosités choriales (8-12 semaines), d'une amniocentèse (14-18 semaines) ou d'une cordocentèse (après la 20e semaine). Dans les échantillons de tissus obtenus du fœtus, l'ensemble de chromosomes est déterminé.
Le Centre de Génétique Moléculaire réalise le diagnostic du syndrome d'Edwards (y compris prénatal) par CF-PCR.
Syndrome d'Edwards (trisomie 18). Causes, symptômes, signes, diagnostic et traitement de la pathologie
Le site fournit Informations d'arrière-plan. Un diagnostic et un traitement adéquats de la maladie sont possibles sous la supervision d'un médecin consciencieux.
 Syndrome d'Edwards ou trisomie 18 est une maladie congénitale grave causée par des anomalies chromosomiques. C'est l'une des pathologies les plus courantes de cette catégorie ( deuxième seulement après le syndrome de Down en fréquence). La maladie se caractérise par de nombreux troubles du développement de divers organes et systèmes. Le pronostic d'un enfant est généralement défavorable, mais beaucoup dépend des soins que les parents sont en mesure de lui prodiguer.
Syndrome d'Edwards ou trisomie 18 est une maladie congénitale grave causée par des anomalies chromosomiques. C'est l'une des pathologies les plus courantes de cette catégorie ( deuxième seulement après le syndrome de Down en fréquence). La maladie se caractérise par de nombreux troubles du développement de divers organes et systèmes. Le pronostic d'un enfant est généralement défavorable, mais beaucoup dépend des soins que les parents sont en mesure de lui prodiguer.
La prévalence du syndrome d'Edwards dans le monde varie de 0,015 à 0,02 %. Il n'y a pas de dépendance claire à l'égard de la localité ou de la race. Statistiquement, les filles tombent malades 3 à 4 fois plus souvent que les garçons. Une explication scientifique de cette proportion n'a pas encore été identifiée. Cependant, un certain nombre de facteurs ont été notés qui peuvent augmenter le risque de cette pathologie.
Comme d'autres mutations chromosomiques, le syndrome d'Edwards est, en principe, une maladie incurable. Plus méthodes modernes le traitement et les soins ne peuvent que maintenir l'enfant en vie et contribuer à certains progrès dans son développement. Il n'y a pas de recommandations uniformes pour la prise en charge de ces enfants en raison de la grande variété de troubles et de complications possibles.
Faits intéressants
- La description des principaux symptômes de cette maladie a été faite au début du XXe siècle.
- Jusqu'au milieu des années 1900, il n'était pas possible de collecter suffisamment d'informations sur cette pathologie. Premièrement, cela nécessitait un niveau de développement technologique approprié qui permettrait la détection d'un chromosome supplémentaire. Deuxièmement, la plupart des enfants meurent dans les premiers jours ou les premières semaines de leur vie en raison du faible niveau de soins médicaux.
- Première Description complète la maladie et sa cause sous-jacente l'apparition d'un 18ème chromosome supplémentaire) n'a été faite qu'en 1960 par le médecin John Edward, d'après qui la nouvelle pathologie a alors été nommée.
- La fréquence réelle du syndrome d'Edwards est de 1 cas pour 2,5 à 3 000 conceptions ( 0,03 – 0,04% ), mais les chiffres officiels sont bien inférieurs. Cela est dû au fait que près de la moitié des embryons présentant cette anomalie ne survivent pas et que la grossesse se termine par un avortement spontané ou une mort intra-utérine du fœtus. Un diagnostic détaillé de la cause d'une fausse couche est rarement effectué.
- La trisomie est une variante d'une mutation chromosomique dans laquelle les cellules d'une personne ne contiennent pas 46, mais 47 chromosomes. Il n'y a que 3 syndromes dans ce groupe de maladies. En plus du syndrome d'Edwards, ce sont les syndromes de Down ( chromosomes de la trisomie 21) et Patau ( trisomie 13 chromosomes). En présence d'autres chromosomes supplémentaires, la pathologie est incompatible avec la vie. Ce n'est que dans ces trois cas qu'il est possible d'avoir un enfant vivant et son autre ( bien que lent) la croissance et le développement.
Causes de la pathologie génétique
 Le syndrome d'Edwards est maladie génétique qui se caractérise par la présence d'un chromosome supplémentaire dans le génome humain. Pour comprendre les raisons qui provoquent les manifestations visibles de cette pathologie, il est nécessaire de savoir ce que sont les chromosomes eux-mêmes et le matériel génétique dans son ensemble.
Le syndrome d'Edwards est maladie génétique qui se caractérise par la présence d'un chromosome supplémentaire dans le génome humain. Pour comprendre les raisons qui provoquent les manifestations visibles de cette pathologie, il est nécessaire de savoir ce que sont les chromosomes eux-mêmes et le matériel génétique dans son ensemble.
Chaque cellule humaine possède un noyau, responsable du stockage et du traitement de l'information génétique. Le noyau contient 46 chromosomes ( 23 couples), qui sont une molécule d'ADN à emballage multiple ( Acide désoxyribonucléique). Cette molécule contient certaines sections appelées gènes. Chaque gène est le prototype d'une protéine spécifique dans le corps humain. Si nécessaire, la cellule lit les informations de ce prototype et produit la protéine appropriée. Les défauts génétiques conduisent à la production de protéines anormales, qui sont responsables de l'apparition de maladies génétiques.
Une paire de chromosomes est constituée de deux molécules d'ADN identiques ( l'un est paternel, l'autre est maternel), qui sont reliés entre eux par un petit pont ( centromère). Le lieu d'adhésion de deux chromosomes dans une paire détermine la forme de l'ensemble de la connexion et son apparence au microscope.
Tous les chromosomes stockent différentes informations génétiques (sur différentes protéines) et sont divisés en groupes suivants :
- groupe A comprend 1 à 3 paires de chromosomes, qui sont grands et en forme de X ;
- groupe B comprend 4 à 5 paires de chromosomes, qui sont également grands, mais le centromère est plus éloigné du centre, c'est pourquoi la forme ressemble à la lettre X avec le centre décalé vers le bas ou vers le haut ;
- groupe C comprend 6 à 12 paires de chromosomes, qui ressemblent aux chromosomes du groupe B en forme, mais leur sont inférieurs en taille;
- groupe D comprend 13 à 15 paires de chromosomes, qui se caractérisent par une taille moyenne et l'emplacement du centromère à la toute fin des molécules, ce qui donne une ressemblance avec la lettre V;
- groupe E comprend 16 à 18 paires de chromosomes, qui se caractérisent par la petite taille et l'emplacement médian du centromère ( Forme en X);
- groupe F comprend 19 à 20 paires de chromosomes, qui sont un peu plus petites que les chromosomes du groupe E et de forme similaire ;
- groupe G comprend 21 à 22 paires de chromosomes, caractérisés par une forme en V et de très petites tailles.
Les 22 paires de chromosomes ci-dessus sont appelées somatiques ou autosomes. De plus, il existe des chromosomes sexuels, qui constituent la 23e paire. Ils ne sont pas similaires en apparence, donc chacun d'eux est désigné séparément. Le chromosome sexuel féminin est désigné X et est similaire au groupe C. Le chromosome sexuel masculin est désigné Y et est similaire en forme et en taille au groupe G. Si l'enfant a les deux chromosomes féminins ( TypeXX), puis une fille est née. Si l'un des chromosomes sexuels est féminin et l'autre masculin, alors un garçon est né ( taper XY). La formule chromosomique s'appelle le caryotype et peut être notée de la manière suivante- 46,XX. Ici, le nombre 46 désigne le nombre total de chromosomes ( 23 couples), et XX est la formule des chromosomes sexuels, qui dépend du sexe ( l'exemple montre le caryotype d'une femme normale).
Le syndrome d'Edwards fait référence aux maladies dites chromosomiques, lorsque le problème n'est pas un défaut génétique, mais un défaut de la molécule d'ADN entière. Pour être plus précis, la forme classique de cette maladie implique la présence d'un 18e chromosome supplémentaire. Le caryotype dans de tels cas est désigné par 47,XX, 18+ ( pour filles) et 47,XY, 18+ ( pour garçon). Le dernier chiffre indique le numéro du chromosome supplémentaire. Un excès d'informations génétiques dans les cellules conduit à l'apparition des manifestations correspondantes de la maladie, qui sont regroupées sous le nom de "syndrome d'Edwards". La présence d'un complément troisième) le chromosome numéro 18 a donné un autre ( plus scientifique) le nom de la maladie est la trisomie 18.
Selon la forme du défaut chromosomique, on distingue trois types de cette maladie:
- Trisomie 18 complète. La forme complète ou classique du syndrome d'Edwards suggère que toutes les cellules du corps ont un chromosome supplémentaire. Cette option la maladie survient dans plus de 90 % des cas et est la plus grave.
- Trisomie partielle 18. La trisomie partielle 18 est un phénomène très rare ( pas plus de 3% de tous les cas de syndrome d'Edwards). Avec lui, les cellules du corps ne contiennent pas un chromosome supplémentaire entier, mais seulement un fragment de celui-ci. Un tel défaut peut être le résultat d'une mauvaise division du matériel génétique, mais il est très rare. Parfois, une partie du dix-huitième chromosome est attachée à une autre molécule d'ADN ( pénètre dans sa structure, allongeant la molécule, ou simplement "s'accroche" à l'aide d'un pont). La division cellulaire ultérieure conduit au fait que le corps a 2 chromosomes normaux numéro 18 et une autre partie des gènes de ces chromosomes ( fragment préservé d'une molécule d'ADN). Dans ce cas, le nombre malformations congénitales sera beaucoup plus faible. Il n'y a pas un excès de toute l'information génétique encodée dans le 18e chromosome, mais seulement une partie de celle-ci. Pour les patients atteints de trisomie 18 partielle, le pronostic est meilleur que pour les enfants avec la forme complète, mais reste toujours défavorable.
- forme de mosaïque. La forme mosaïque du syndrome d'Edwards survient dans 5 à 7% des cas de cette maladie. Le mécanisme de son apparition diffère des autres espèces. Le fait est qu'ici le défaut s'est formé après la fusion du spermatozoïde et de l'ovule. Les deux gamètes ( cellules sexuelles) avait initialement un caryotype normal et portait un chromosome de chaque espèce. Après la fusion, une cellule de formule normale 46,XX ou 46,XY s'est formée. Lors du processus de division de cette cellule, une panne s'est produite. Lors du doublement du matériel génétique, l'un des fragments a reçu un 18e chromosome supplémentaire. Ainsi, à un certain stade, un embryon s'est formé, dont certaines cellules ont un caryotype normal ( par exemple 46,XX), et une partie est le caryotype du syndrome d'Edwards ( 47,XX, 18+). La proportion de cellules pathologiques ne dépasse jamais 50 %. Leur nombre dépend du stade de division de la cellule initiale où la panne s'est produite. Plus cela se produit tard, plus la proportion de cellules défectueuses sera faible. La forme tire son nom du fait que toutes les cellules du corps forment une sorte de mosaïque. Certains d'entre eux sont en bonne santé et certains ont une pathologie génétique grave. Dans le même temps, il n'y a pas de modèles dans la distribution des cellules dans le corps, c'est-à-dire que toutes les cellules défectueuses ne peuvent pas être localisées à un seul endroit pour pouvoir être éliminées. L'état général du patient est plus facile qu'avec la forme classique de trisomie 18.
La présence d'un chromosome supplémentaire dans le génome humain pose de nombreux problèmes. Le fait est que les cellules humaines sont programmées pour lire l'information génétique et ne dupliquer que le nombre de molécules d'ADN donné par la nature. Des violations, même dans la structure d'un gène, peuvent entraîner des maladies graves. En présence d'une molécule d'ADN entière, de multiples troubles se développent même au stade du développement intra-utérin avant la naissance d'un enfant.
Selon des études récentes, le chromosome numéro 18 contient 557 gènes qui codent pour au moins 289 protéines différentes. En termes de pourcentage, cela représente environ 2,5 % du matériel génétique total. Les perturbations qu'un tel déséquilibre provoque sont très graves. Une quantité incorrecte de protéines prédétermine de nombreuses anomalies dans le développement de divers organes et tissus. Dans le cas du syndrome d'Edwards, les os du crâne, certaines parties du système nerveux, les systèmes cardiovasculaire et génito-urinaire souffrent plus souvent que d'autres. Apparemment, cela est dû au fait que les gènes situés sur ce chromosome sont liés au développement de ces organes et systèmes.
Ainsi, la principale et unique cause du syndrome d'Edwards est la présence d'une molécule d'ADN supplémentaire. le plus souvent ( dans la forme classique de la maladie) est hérité de l'un des parents. Normalement, chaque gamète ( sperme et ovule) contiennent 22 chromosomes somatiques non appariés, plus un chromosome sexuel. Une femme envoie toujours à un enfant un ensemble standard de 22+X, et un homme peut envoyer 22+X ou 22+Y. Cela détermine le sexe de l'enfant. Les cellules germinales des parents sont formées à la suite de la division de cellules ordinaires en deux ensembles. Normalement, la cellule mère se divise en deux parties égales, mais parfois tous les chromosomes ne se divisent pas en deux. Si la 18e paire ne s'est pas dispersée le long des pôles de la cellule, alors l'un des œufs ( ou l'un des spermatozoïdes) sera défectueux à l'avance. Il n'aura pas 23, mais 24 chromosomes. Si c'est cette cellule qui participe à la fécondation, l'enfant recevra un 18e chromosome supplémentaire.
Les facteurs suivants peuvent affecter une mauvaise division cellulaire :
- Âge des parents. Il a été prouvé que la probabilité d'anomalies chromosomiques augmente en proportion directe avec l'âge de la mère. Dans le syndrome d'Edwards, cette relation est moins prononcée que dans d'autres pathologies similaires ( par exemple le syndrome de Down). Mais pour les femmes de plus de 40 ans, le risque d'avoir un enfant atteint de cette pathologie est en moyenne 6 à 7 fois plus élevé. Une dépendance similaire à l'âge du père est observée dans une bien moindre mesure.
- Tabagisme et alcool. De mauvaises habitudes telles que le tabagisme et l'abus d'alcool peuvent affecter le système reproducteur humain, affectant la division des cellules germinales. Ainsi, l'utilisation régulière de ces substances ( ainsi que d'autres médicaments) augmente le risque de mauvaise affectation du matériel génétique.
- Réception médicaments . Quelques médicaments s'ils sont mal pris au cours du premier trimestre, ils peuvent affecter la division des cellules germinales et provoquer une forme mosaïque du syndrome d'Edwards.
- Maladies de la région génitale. Les infections passées avec des dommages aux organes reproducteurs peuvent affecter la division correcte des cellules. Ils augmentent le risque de troubles chromosomiques et génétiques en général, bien que de telles études n'aient pas été menées spécifiquement pour le syndrome d'Edwards.
- rayonnement rayonnement. L'exposition des organes génitaux aux rayons X ou à d'autres rayonnements ionisants peut provoquer des mutations génétiques. Une telle influence externe est particulièrement dangereuse à l'adolescence, lorsque la division cellulaire est la plus active. Les particules qui forment le rayonnement pénètrent facilement dans les tissus et exposent la molécule d'ADN à une sorte de « bombardement ». Si cela se produit au moment de la division cellulaire, le risque de mutation chromosomique est particulièrement élevé.
De manière générale, on ne peut pas dire que les causes du développement du syndrome d'Edwards soient enfin connues et bien étudiées. Les facteurs ci-dessus ne font qu'augmenter le risque de développer cette mutation. La prédisposition congénitale de certaines personnes à une distribution incorrecte du matériel génétique dans les cellules germinales n'est pas exclue. Par exemple, on pense que dans un couple marié qui a déjà donné naissance à un enfant atteint du syndrome d'Edwards, la probabilité d'avoir un deuxième enfant avec une pathologie similaire est de 2 à 3% ( environ 200 fois plus élevée que la prévalence moyenne de cette maladie).
À quoi ressemblent les nouveau-nés atteints du syndrome d'Edwards ?
 Comme vous le savez, le syndrome d'Edwards peut être diagnostiqué avant la naissance, mais dans la plupart des cas, cette maladie est détectée immédiatement après la naissance d'un enfant. Les nouveau-nés atteints de cette pathologie présentent un certain nombre d'anomalies de développement prononcées, qui permettent parfois de suspecter immédiatement le diagnostic correct. La confirmation est effectuée plus tard à l'aide d'une analyse génétique spéciale.
Comme vous le savez, le syndrome d'Edwards peut être diagnostiqué avant la naissance, mais dans la plupart des cas, cette maladie est détectée immédiatement après la naissance d'un enfant. Les nouveau-nés atteints de cette pathologie présentent un certain nombre d'anomalies de développement prononcées, qui permettent parfois de suspecter immédiatement le diagnostic correct. La confirmation est effectuée plus tard à l'aide d'une analyse génétique spéciale.
Les nouveau-nés atteints du syndrome d'Edwards présentent les anomalies développementales caractéristiques suivantes :
- changement de forme du crâne;
- changement de forme des oreilles;
- anomalies dans le développement du ciel;
- chaise berçante pour les pieds;
- longueur anormale des doigts;
- changement de forme de la mâchoire inférieure;
- fusion des doigts;
- anomalies dans le développement des organes génitaux;
- position fléchisseur des mains;
- caractéristiques dermatoglyphiques.
Changer la forme du crâne
 Un symptôme typique du syndrome d'Edwards est la dolichocéphalie. C'est le nom d'un changement caractéristique de la forme de la tête d'un nouveau-né, qui se produit également dans certaines autres maladies génétiques. Chez les dolichocéphales ( enfants avec ce symptôme) un crâne plus long et plus étroit. La présence de cette anomalie est précisément confirmée par des mesures particulières. Déterminer le rapport de la largeur du crâne au niveau des os pariétaux à la longueur du crâne ( de la saillie au-dessus du pont du nez à l'occiput). Si le ratio obtenu est inférieur à 75 %, alors cet enfant fait référence aux dolichocéphales. En soi, ce symptôme n'est pas une violation grave. Ce n'est qu'un des types de forme de crâne que l'on trouve également dans absolument personnes normales. Les enfants atteints du syndrome d'Edwards dans 80 à 85% des cas sont dolichocéphales prononcés, dans lesquels la disproportion dans la longueur et la largeur du crâne peut être observée même sans mesures spéciales.
Un symptôme typique du syndrome d'Edwards est la dolichocéphalie. C'est le nom d'un changement caractéristique de la forme de la tête d'un nouveau-né, qui se produit également dans certaines autres maladies génétiques. Chez les dolichocéphales ( enfants avec ce symptôme) un crâne plus long et plus étroit. La présence de cette anomalie est précisément confirmée par des mesures particulières. Déterminer le rapport de la largeur du crâne au niveau des os pariétaux à la longueur du crâne ( de la saillie au-dessus du pont du nez à l'occiput). Si le ratio obtenu est inférieur à 75 %, alors cet enfant fait référence aux dolichocéphales. En soi, ce symptôme n'est pas une violation grave. Ce n'est qu'un des types de forme de crâne que l'on trouve également dans absolument personnes normales. Les enfants atteints du syndrome d'Edwards dans 80 à 85% des cas sont dolichocéphales prononcés, dans lesquels la disproportion dans la longueur et la largeur du crâne peut être observée même sans mesures spéciales.
Une autre variante d'une anomalie dans le développement du crâne est la soi-disant microcéphalie, dans laquelle la taille de la tête dans son ensemble est trop petite par rapport au reste du corps. Tout d'abord, cela ne s'applique pas au crâne facial ( mâchoires, pommettes, orbites), à savoir le crâne, dans lequel se trouve le cerveau. La microcéphalie est moins caractéristique du syndrome d'Edwards que la dolichocéphalie, mais elle survient également avec une fréquence plus élevée que chez les personnes en bonne santé.
Changer la forme de l'oreille
 Si la dolichocéphalie peut être une variante de la norme, la pathologie du développement de l'oreillette chez les enfants atteints du syndrome d'Edwards est beaucoup plus grave. Dans une certaine mesure, ce symptôme est observé chez plus de 95% des enfants atteints de la forme complète de cette maladie. Avec une forme de mosaïque, sa fréquence est un peu moindre. L'oreillette est généralement située plus bas que chez les personnes normales ( parfois sous le niveau des yeux). Les renflements caractéristiques du cartilage qui forme l'oreillette sont mal définis ou absents. Le lobe de l'oreille ou le tragus peuvent également être absents ( une petite zone saillante de cartilage devant le conduit auditif). Le conduit auditif lui-même est généralement rétréci et dans environ 20 à 25%, il est complètement absent.
Si la dolichocéphalie peut être une variante de la norme, la pathologie du développement de l'oreillette chez les enfants atteints du syndrome d'Edwards est beaucoup plus grave. Dans une certaine mesure, ce symptôme est observé chez plus de 95% des enfants atteints de la forme complète de cette maladie. Avec une forme de mosaïque, sa fréquence est un peu moindre. L'oreillette est généralement située plus bas que chez les personnes normales ( parfois sous le niveau des yeux). Les renflements caractéristiques du cartilage qui forme l'oreillette sont mal définis ou absents. Le lobe de l'oreille ou le tragus peuvent également être absents ( une petite zone saillante de cartilage devant le conduit auditif). Le conduit auditif lui-même est généralement rétréci et dans environ 20 à 25%, il est complètement absent.
Anomalies dans le développement du ciel
 Les processus palatins de la mâchoire supérieure fusionnent au cours du développement de l'embryon, formant un palais dur. Chez les enfants atteints du syndrome d'Edwards, ce processus reste souvent incomplet. À l'endroit où se trouve la suture médiane chez les personnes normales ( on peut le sentir au milieu du palais dur avec la langue) ils présentent un espace longitudinal.
Les processus palatins de la mâchoire supérieure fusionnent au cours du développement de l'embryon, formant un palais dur. Chez les enfants atteints du syndrome d'Edwards, ce processus reste souvent incomplet. À l'endroit où se trouve la suture médiane chez les personnes normales ( on peut le sentir au milieu du palais dur avec la langue) ils présentent un espace longitudinal.
Il existe plusieurs variantes de ce défaut :
- non-occlusion du palais mou ( dos, partie profonde du palais qui surplombe le pharynx);
- non-fermeture partielle du palais dur ( l'écart ne s'étend pas sur toute la mâchoire supérieure);
- non-fermeture complète du palais dur et mou;
- non-fermeture complète du palais et des lèvres.
Dans certains cas, le dédoublement du ciel est bilatéral. Deux coins saillants de la lèvre supérieure sont le début de fissures pathologiques. L'enfant ne peut pas fermer complètement la bouche à cause de ce défaut. Dans les cas graves, la communication des cavités buccale et nasale est clairement visible ( même la bouche fermée). Les dents antérieures peuvent manquer ou pousser sur le côté à l'avenir.
Ces défauts de développement sont également connus sous le nom de fente palatine, fente palatine et fente labiale. Tous peuvent survenir en dehors du syndrome d'Edwards, cependant, chez les enfants atteints de cette pathologie, leur fréquence est particulièrement élevée ( près de 20 % des nouveau-nés). Beaucoup plus fréquemment ( jusqu'à 65% des nouveau-nés) ont une caractéristique différente connue sous le nom de ciel haut ou gothique. Il peut être attribué aux variantes de la norme, car on le trouve également chez les personnes en bonne santé.
La présence d'une fente palatine ou de la lèvre supérieure ne confirme pas encore le syndrome d'Edwards. Cette malformation peut survenir avec une fréquence assez élevée et indépendamment sans troubles concomitants d'autres organes et systèmes. Il existe un certain nombre d'interventions chirurgicales standard pour corriger cette anomalie.
Pied basculant
 C'est le nom d'un changement caractéristique du pied, qui survient principalement dans le cadre du syndrome d'Edwards. Sa fréquence dans cette maladie atteint 75 %. Le défaut réside dans la position incorrecte des os du talus, du calcanéum et du scaphoïde. Il appartient à la catégorie des déformations en valgus plat du pied chez l'enfant.
C'est le nom d'un changement caractéristique du pied, qui survient principalement dans le cadre du syndrome d'Edwards. Sa fréquence dans cette maladie atteint 75 %. Le défaut réside dans la position incorrecte des os du talus, du calcanéum et du scaphoïde. Il appartient à la catégorie des déformations en valgus plat du pied chez l'enfant.
Extérieurement, le pied d'un nouveau-né ressemble à ceci. Le tubercule calcanéen, sur lequel repose l'arrière du pied, fait saillie vers l'arrière. Dans ce cas, le coffre-fort peut être complètement absent. Ceci est facile à voir en regardant le pied de l'intérieur. Normalement, une ligne concave y apparaît, allant du talon à la base du gros orteil. Avec un stop à bascule, cette ligne est absente. Le pied est plat voire convexe. Cela lui donne une ressemblance avec les pieds d'une chaise berçante.
Longueur de doigt anormale
 Chez les enfants atteints du syndrome d'Edwards, une proportion anormale de la longueur des orteils peut être observée dans le contexte de modifications de la structure du pied. En particulier, nous parlons du pouce, qui est normalement le plus long. Chez les nouveau-nés atteints de ce syndrome, il est inférieur en longueur au deuxième doigt. Ce défaut ne peut être vu qu'en redressant les doigts et en les examinant attentivement. Avec l'âge, à mesure que l'enfant grandit, cela devient plus visible. Étant donné que le raccourcissement du gros orteil se produit principalement avec le pied basculant, la prévalence de ces symptômes chez les nouveau-nés est à peu près la même.
Chez les enfants atteints du syndrome d'Edwards, une proportion anormale de la longueur des orteils peut être observée dans le contexte de modifications de la structure du pied. En particulier, nous parlons du pouce, qui est normalement le plus long. Chez les nouveau-nés atteints de ce syndrome, il est inférieur en longueur au deuxième doigt. Ce défaut ne peut être vu qu'en redressant les doigts et en les examinant attentivement. Avec l'âge, à mesure que l'enfant grandit, cela devient plus visible. Étant donné que le raccourcissement du gros orteil se produit principalement avec le pied basculant, la prévalence de ces symptômes chez les nouveau-nés est à peu près la même.
Chez l'adulte, le raccourcissement du gros orteil n'a pas une telle valeur diagnostique. Un tel défaut peut être une caractéristique individuelle chez une personne en bonne santé ou une conséquence d'autres facteurs ( déformation articulaire, maladie osseuse, port de chaussures mal ajustées). À cet égard, ce signe doit être considéré comme un symptôme possible uniquement chez les nouveau-nés en présence d'autres anomalies du développement.
Changer la forme de la mâchoire inférieure
 Des modifications de la forme de la mâchoire inférieure chez les nouveau-nés surviennent dans près de 70% des cas. Normalement, le menton chez les enfants ne dépasse pas vers l'avant comme chez les adultes, mais chez les patients atteints du syndrome d'Edwards, il est trop rétracté. Cela est dû au sous-développement de la mâchoire inférieure, appelée micrognathie ( microgénie). Ce symptôme se retrouve également dans d'autres maladies congénitales. Il n'est pas rare de trouver des adultes avec des traits faciaux similaires. En l'absence de pathologies concomitantes, ceci est considéré comme une variante de la norme, bien qu'il entraîne certaines difficultés.
Des modifications de la forme de la mâchoire inférieure chez les nouveau-nés surviennent dans près de 70% des cas. Normalement, le menton chez les enfants ne dépasse pas vers l'avant comme chez les adultes, mais chez les patients atteints du syndrome d'Edwards, il est trop rétracté. Cela est dû au sous-développement de la mâchoire inférieure, appelée micrognathie ( microgénie). Ce symptôme se retrouve également dans d'autres maladies congénitales. Il n'est pas rare de trouver des adultes avec des traits faciaux similaires. En l'absence de pathologies concomitantes, ceci est considéré comme une variante de la norme, bien qu'il entraîne certaines difficultés.
Les nouveau-nés atteints de micrognathie développent généralement rapidement les problèmes suivants :
- incapacité à garder la bouche fermée pendant longtemps ( baver);
- difficultés d'alimentation;
- développement tardif des dents et leur emplacement incorrect.
Espace entre le bas et mâchoire supérieure peut dépasser 1 cm, ce qui est beaucoup compte tenu de la taille de la tête du bébé.
Fusion des doigts
 La fusion des doigts, ou scientifiquement syndactylie, se produit chez environ 45 % des nouveau-nés. Le plus souvent, cette anomalie touche les orteils, mais on retrouve également une syndactylie au niveau des mains. Dans les cas bénins, la fusion est formée par un pli cutané comme une courte membrane. Dans les cas plus graves, une fusion avec des ponts de tissu osseux est observée.
La fusion des doigts, ou scientifiquement syndactylie, se produit chez environ 45 % des nouveau-nés. Le plus souvent, cette anomalie touche les orteils, mais on retrouve également une syndactylie au niveau des mains. Dans les cas bénins, la fusion est formée par un pli cutané comme une courte membrane. Dans les cas plus graves, une fusion avec des ponts de tissu osseux est observée.
La syndactylie survient non seulement dans le syndrome d'Edwards, mais également dans de nombreuses autres maladies chromosomiques. Il y a aussi des cas où cette malformation était la seule, et sinon le patient ne différait en rien des enfants normaux. À cet égard, la fusion des doigts n'est qu'un des signes possibles du syndrome d'Edwards, ce qui permet de suspecter le diagnostic, mais ne le confirme pas.
Anomalies dans le développement des organes génitaux
Position fléchisseur des mains
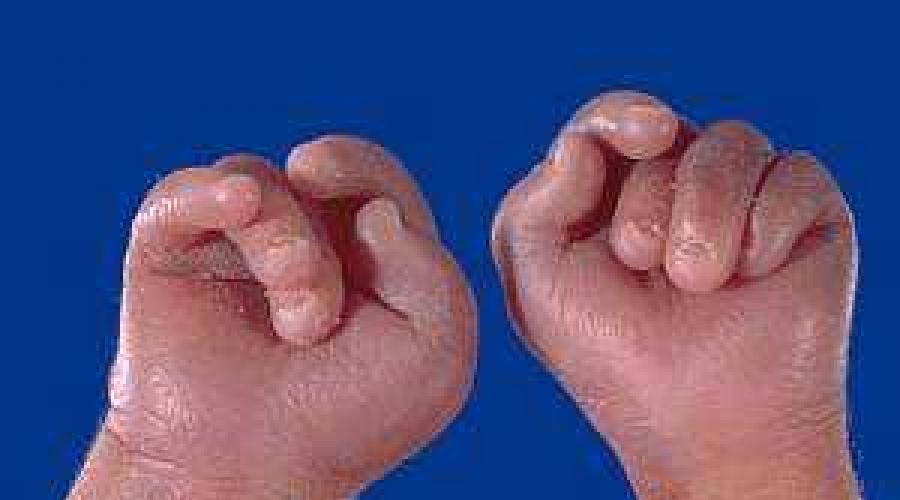
 La position des fléchisseurs des mains est une disposition spéciale des doigts, causée non pas tant par des troubles structurels dans la région de la main que par une augmentation du tonus musculaire. Les fléchisseurs des doigts et des mains sont constamment tendus, c'est pourquoi le pouce et l'auriculaire semblent recouvrir le reste des doigts, qui sont pressés contre la paume. Ce symptôme est observé dans de nombreuses pathologies congénitales et n'est pas caractéristique du syndrome d'Edwards. Cependant, si une brosse de forme similaire est trouvée, cette pathologie doit être supposée. Avec lui, la position du fléchisseur des doigts est observée chez près de 90% des nouveau-nés.
La position des fléchisseurs des mains est une disposition spéciale des doigts, causée non pas tant par des troubles structurels dans la région de la main que par une augmentation du tonus musculaire. Les fléchisseurs des doigts et des mains sont constamment tendus, c'est pourquoi le pouce et l'auriculaire semblent recouvrir le reste des doigts, qui sont pressés contre la paume. Ce symptôme est observé dans de nombreuses pathologies congénitales et n'est pas caractéristique du syndrome d'Edwards. Cependant, si une brosse de forme similaire est trouvée, cette pathologie doit être supposée. Avec lui, la position du fléchisseur des doigts est observée chez près de 90% des nouveau-nés.
Caractéristiques dermatoglyphiques
 Avec de nombreuses anomalies chromosomiques, les nouveau-nés présentent des changements dermatoglyphiques caractéristiques ( motifs anormaux et plis sur la peau des paumes). Avec le syndrome d'Edwards, certains signes peuvent être retrouvés dans près de 60% des cas. Ils sont importants principalement pour le diagnostic préliminaire en cas de mosaïque ou de forme partielle de la maladie. Avec une trisomie 18 complète, les dermatoglyphes ne sont pas utilisés, car il existe suffisamment d'autres anomalies de développement plus visibles pour suspecter le syndrome d'Edwards.
Avec de nombreuses anomalies chromosomiques, les nouveau-nés présentent des changements dermatoglyphiques caractéristiques ( motifs anormaux et plis sur la peau des paumes). Avec le syndrome d'Edwards, certains signes peuvent être retrouvés dans près de 60% des cas. Ils sont importants principalement pour le diagnostic préliminaire en cas de mosaïque ou de forme partielle de la maladie. Avec une trisomie 18 complète, les dermatoglyphes ne sont pas utilisés, car il existe suffisamment d'autres anomalies de développement plus visibles pour suspecter le syndrome d'Edwards.
Les principales caractéristiques dermatoglyphiques du syndrome d'Edwards sont :
- les arcs au bout des doigts sont localisés plus fréquemment que chez les personnes en bonne santé;
- pli cutané entre le dernier ( clou) et avant-dernier ( milieu) les phalanges des doigts sont absentes ;
- 30% des nouveau-nés ont une rainure dite transversale dans la paume ( ligne de singe, ligne simienne).
Des études spéciales peuvent révéler d'autres écarts par rapport à la norme, mais immédiatement après la naissance, sans la participation de spécialistes étroits, ces changements suffisent aux médecins.
En plus des signes ci-dessus, il existe un certain nombre d'anomalies de développement possibles qui peuvent aider au diagnostic préliminaire du syndrome d'Edwards. Selon certaines données, avec un examen externe détaillé, jusqu'à 50 signes externes peuvent être détectés. La combinaison des plus symptômes courants, présenté ci-dessus, indique avec une forte probabilité la présence de cette pathologie grave chez l'enfant. Avec une variante en mosaïque du syndrome d'Edwards, il peut ne pas y avoir plusieurs anomalies, mais la présence d'une seule d'entre elles est une indication pour un test génétique spécial.
À quoi ressemblent les enfants atteints du syndrome d'Edwards ?
 Les enfants atteints du syndrome d'Edwards développent généralement une variété de comorbidités en vieillissant. Leurs symptômes commencent à apparaître quelques semaines après la naissance. Ces symptômes peuvent être la première manifestation du syndrome, car avec une variante mosaïque, dans de rares cas, la maladie peut passer inaperçue immédiatement après la naissance. Ensuite, le diagnostic de la maladie devient plus compliqué.
Les enfants atteints du syndrome d'Edwards développent généralement une variété de comorbidités en vieillissant. Leurs symptômes commencent à apparaître quelques semaines après la naissance. Ces symptômes peuvent être la première manifestation du syndrome, car avec une variante mosaïque, dans de rares cas, la maladie peut passer inaperçue immédiatement après la naissance. Ensuite, le diagnostic de la maladie devient plus compliqué.
La plupart des manifestations extérieures du syndrome observées à la naissance persistent et deviennent plus perceptibles. Nous parlons de la forme du crâne, du pied basculant, de la déformation de l'oreillette, etc. Peu à peu, d'autres manifestations externes commencent à s'ajouter qui ne pouvaient pas être remarquées immédiatement après la naissance. Dans ce cas, nous parlons de signes pouvant apparaître chez les enfants au cours de la première année de vie.
Les enfants atteints du syndrome d'Edwards ont les caractéristiques externes suivantes :
- arriéré Développement physique;
- pied bot;
- tonus musculaire anormal;
- réactions émotionnelles anormales.
Retard dans le développement physique
Le retard de développement physique s'explique par le faible poids corporel de l'enfant à la naissance ( seulement 2000 - 2200 g à un âge gestationnel normal). Un défaut génétique joue également un rôle important, ce qui ne permet pas à tous les systèmes de l'organisme de se développer normalement et harmonieusement. Les principaux indicateurs permettant d'évaluer la croissance et le développement de l'enfant sont considérablement réduits.
Vous pouvez remarquer l'arriéré d'un enfant par les indicateurs anthropométriques suivants :
- taille de l'enfant ;
- poids de l'enfant;
- Tour de poitrine;
- Circonférence de la tête ( cet indicateur peut être normal ou même augmenté, mais on ne peut pas s'y fier en raison d'une déformation congénitale du crâne).
Pied bot
Tonus musculaire anormal
Réactions émotionnelles anormales
À quoi ressemblent les adultes atteints du syndrome d'Edwards ?
 Dans la grande majorité des cas, les enfants nés avec le syndrome d'Edwards ne survivent pas jusqu'à l'âge adulte. Dans la forme complète de cette maladie, lorsqu'un chromosome supplémentaire est présent dans chaque cellule du corps, 90 % des enfants meurent avant l'âge de 1 an en raison d'anomalies graves dans le développement des organes internes. Même avec la correction chirurgicale des éventuels défauts et des soins de qualité, leur corps est plus sensible aux maladies infectieuses. Ceci est facilité par les troubles de l'alimentation qui surviennent chez la plupart des enfants. Tout cela explique la mortalité la plus élevée dans le syndrome d'Edwards.
Dans la grande majorité des cas, les enfants nés avec le syndrome d'Edwards ne survivent pas jusqu'à l'âge adulte. Dans la forme complète de cette maladie, lorsqu'un chromosome supplémentaire est présent dans chaque cellule du corps, 90 % des enfants meurent avant l'âge de 1 an en raison d'anomalies graves dans le développement des organes internes. Même avec la correction chirurgicale des éventuels défauts et des soins de qualité, leur corps est plus sensible aux maladies infectieuses. Ceci est facilité par les troubles de l'alimentation qui surviennent chez la plupart des enfants. Tout cela explique la mortalité la plus élevée dans le syndrome d'Edwards.
Avec une forme de mosaïque plus douce, lorsque seule une fraction des cellules du corps contient un ensemble anormal de chromosomes, le taux de survie est un peu plus élevé. Cependant, même dans ces cas, seuls quelques patients survivent jusqu'à l'âge adulte. Leur apparence défini par des anomalies congénitales présentes à la naissance ( fente labiale, oreillette déformée, etc.). Le principal symptôme, présent chez tous les enfants sans exception, est un retard mental grave. Ayant vécu jusqu'à l'âge adulte, un enfant atteint du syndrome d'Edwards est un oligophrène profond ( QI inférieur à 20, ce qui correspond au degré de retard mental le plus sévère). En général, des cas isolés sont décrits dans la littérature médicale lorsque des enfants atteints du syndrome d'Edwards ont survécu jusqu'à l'âge adulte. De ce fait, trop peu de données objectives ont été accumulées pour parler des signes extérieurs de cette maladie chez l'adulte.
Diagnostic de pathologie génétique
 Actuellement, il existe trois étapes principales dans le diagnostic du syndrome d'Edwards, chacune comprenant plusieurs méthodes possibles. Étant donné que cette maladie est incurable, les parents doivent prêter attention aux possibilités de ces méthodes et les utiliser. La plupart des tests sont effectués dans des centres spécialisés pour le diagnostic prénatal, où il y a tout l'équipement nécessaire pour rechercher des maladies génétiques. Cependant, même une consultation avec un généticien ou un néonatologiste peut être utile.
Actuellement, il existe trois étapes principales dans le diagnostic du syndrome d'Edwards, chacune comprenant plusieurs méthodes possibles. Étant donné que cette maladie est incurable, les parents doivent prêter attention aux possibilités de ces méthodes et les utiliser. La plupart des tests sont effectués dans des centres spécialisés pour le diagnostic prénatal, où il y a tout l'équipement nécessaire pour rechercher des maladies génétiques. Cependant, même une consultation avec un généticien ou un néonatologiste peut être utile.
Le diagnostic du syndrome d'Edwards est possible aux stades suivants :
- diagnostic avant la conception;
- diagnostic pendant le développement fœtal;
- diagnostic après la naissance.
Diagnostic avant la conception
Le diagnostic avant la conception d'un enfant est une option idéale, mais malheureusement, au stade actuel de développement de la médecine, ses possibilités sont très limitées. Les médecins peuvent utiliser plusieurs méthodes pour suggérer un risque accru d'avoir un enfant atteint d'une anomalie chromosomique, mais pas plus. Le fait est qu'avec le syndrome d'Edwards, en principe, les violations chez les parents ne peuvent pas être détectées. Une cellule sexuelle défectueuse avec 24 chromosomes n'est qu'une parmi plusieurs milliers. Par conséquent, il est impossible de dire avec certitude jusqu'au moment de la conception si un enfant naîtra avec cette maladie.
Les principales méthodes de diagnostic avant la conception sont:
- Histoire de famille. Une histoire familiale est un questionnement détaillé des deux parents sur leur ascendance. Le médecin s'intéresse à tous les cas d'hérédité ( et surtout chromosomique) maladies dans la famille. Si au moins un des parents se souvient d'un cas de trisomie ( Syndrome d'Edwards, syndrome de Down, Patau), ce qui augmente considérablement la probabilité d'avoir un enfant malade. Cependant, le risque est toujours inférieur à 1 %. Avec des cas répétés de ces maladies chez les ancêtres, le risque augmente plusieurs fois. En fait, l'analyse se résume à une consultation avec un néonatologiste ou un généticien. Auparavant, les parents pouvaient essayer de collecter des informations plus détaillées sur leurs ancêtres ( de préférence 3-4 genoux). Cela améliorera la précision de cette méthode.
- Détection des facteurs de risque. Le principal facteur de risque qui augmente objectivement le risque d'anomalies chromosomiques est l'âge de la mère. Comme mentionné ci-dessus, chez les mères après 40 ans, la probabilité d'avoir un enfant atteint du syndrome d'Edwards augmente plusieurs fois. Selon certains rapports, après 45 ans ( âge de la mère) presque une grossesse sur cinq s'accompagne d'une pathologie chromosomique. La plupart d'entre eux se terminent par une fausse couche. D'autres facteurs sont les maladies infectieuses passées, les maladies chroniques, les mauvaises habitudes. Cependant, leur rôle dans le diagnostic est beaucoup plus faible. Cette méthode ne donne pas non plus de réponse exacte à la question de savoir si un enfant atteint du syndrome d'Edwards sera conçu.
- Analyse génétique des parents. Si les méthodes précédentes se limitaient à interroger les parents, l'analyse génétique est une étude complète qui nécessite un équipement spécial, des réactifs et des spécialistes qualifiés. Le sang est prélevé sur les parents, à partir duquel les leucocytes sont isolés en laboratoire. Après traitement avec des substances spéciales dans ces cellules, les chromosomes au stade de la division deviennent clairement visibles. Ainsi, le caryotype des parents est compilé. Dans la plupart des cas, c'est normal avec des troubles chromosomiques que l'on peut trouver ici, la probabilité de procréation est négligeable). De plus, à l'aide de marqueurs spéciaux ( fragments de chaînes moléculaires) il est possible de détecter des sections d'ADN avec des gènes défectueux. Cependant, on ne trouvera pas ici d'anomalies chromosomiques, mais des mutations génétiques qui n'affectent pas directement la probabilité de syndrome d'Edwards. Ainsi, l'analyse génétique des parents avant le moment de la conception, malgré la complexité et le coût élevé, ne donne pas non plus de réponse sans ambiguïté concernant le pronostic de cette pathologie.
Diagnostic au cours du développement fœtal
Au cours du développement fœtal, plusieurs moyens permettent de confirmer directement ou indirectement la présence d'une pathologie chromosomique dans l'embryon. La précision de ces méthodes est beaucoup plus élevée, car les médecins ne traitent pas avec les parents, mais avec le fœtus lui-même. L'embryon lui-même et ses cellules avec leur propre ADN sont disponibles pour étude. Cette étape est également appelée diagnostic prénatal et est la plus importante. À ce stade, vous pouvez confirmer le diagnostic, avertir les parents de la présence d'une pathologie et, si nécessaire, interrompre la grossesse. Si la femme décide d'accoucher et que le nouveau-né est vivant, les médecins pourront alors se préparer à l'avance pour lui fournir l'assistance nécessaire.
Les principales méthodes de recherche dans le cadre du diagnostic prénatal sont :
- Procédure d'échographie ( ultrason) . Cette méthode est non invasive, c'est-à-dire qu'elle n'endommage pas les tissus de la mère ou du fœtus. Il est totalement sans danger et est recommandé à toutes les femmes enceintes dans le cadre du diagnostic prénatal ( quel que soit leur âge ou leur risque accru de troubles chromosomiques). Le programme standard suggère que l'échographie doit être effectuée trois fois ( à 10 - 14, 20 - 24 et 32 - 34 semaines de grossesse). Si le médecin traitant suppose la possibilité de malformations congénitales, une échographie non planifiée peut également être effectuée. Le syndrome d'Edwards peut être indiqué par le retard du fœtus en taille et en poids, une grande quantité de liquide amniotique, des anomalies de développement visibles ( microcéphalie, déformation osseuse). Ces troubles sont très susceptibles d'indiquer des maladies génétiques graves, mais le syndrome d'Edwards ne peut pas être définitivement confirmé.
- Amniocentèse. L'amniocentèse est une cytologie ( cellulaire) analyse du liquide amniotique. Le médecin insère doucement une aiguille spéciale sous le contrôle d'un échographe. La ponction est faite dans un endroit où il n'y a pas de boucles du cordon ombilical. À l'aide d'une seringue, la quantité de liquide amniotique nécessaire à l'étude est prélevée. La procédure peut être effectuée à tous les trimestres de la grossesse, mais le moment optimal pour le diagnostic des troubles chromosomiques est la période après la 15e semaine de grossesse. Taux de complications ( jusqu'à l'avortement spontané) est jusqu'à 1%, de sorte que la procédure ne doit pas être effectuée en l'absence de toute indication. Une fois le liquide amniotique prélevé, le matériau obtenu est traité. Ils contiennent des cellules liquides de la surface de la peau du bébé, qui contiennent des échantillons de son ADN. Ce sont eux qui sont testés pour la présence de maladies génétiques.
- Cordocentèse. La cordocentèse est la méthode la plus informative de diagnostic prénatal. Après anesthésie et sous le contrôle d'un appareil à ultrasons, le médecin perce un vaisseau traversant le cordon ombilical avec une aiguille spéciale. Ainsi, un échantillon de sang est obtenu ( jusqu'à 5 ml) d'un enfant en développement. La technique d'analyse est similaire à celle des adultes. Ce matériel peut être examiné avec une grande précision pour diverses anomalies génétiques. Cela inclut le caryotype fœtal. En présence d'un 18e chromosome supplémentaire, on peut parler de syndrome d'Edwards confirmé. Cette analyse est recommandée après la 18ème semaine de grossesse ( optimale 22 - 25 semaines). La fréquence des complications possibles après cordocentèse est de 1,5 à 2%.
- Biopsie chorionique. Le chorion est l'une des membranes germinales contenant des cellules avec l'information génétique du fœtus. Cette étude implique la ponction de l'utérus sous anesthésie à travers la paroi abdominale antérieure. À l'aide d'une pince à biopsie spéciale, un échantillon de tissu est prélevé pour analyse. Ensuite, une étude génétique standard du matériel obtenu est effectuée. Le caryotypage est effectué pour diagnostiquer le syndrome d'Edwards. Le moment optimal pour une biopsie du chorion est considéré comme 9-12 semaines de grossesse. La fréquence des complications est de 2 à 3 %. Le principal avantage qui la distingue des autres méthodes est la rapidité d'obtention du résultat ( sous 2 à 4 jours).
Diagnostic après la naissance
Le diagnostic du syndrome d'Edwards après la naissance est le plus simple, le plus rapide et le plus précis. Malheureusement, à ce stade, un enfant atteint d'une pathologie génétique sévère est déjà né, traitement efficace qui n'existe pas actuellement. Si la maladie n'a pas été détectée au stade du diagnostic prénatal ( ou des études pertinentes n'ont pas été menées), la suspicion de syndrome d'Edwards apparaît immédiatement après la naissance. L'enfant est généralement à terme ou même post-terme, mais son poids est toujours inférieur à la moyenne. De plus, certaines des malformations congénitales mentionnées ci-dessus attirent l'attention. S'ils sont remarqués, une analyse génétique est effectuée pour confirmer le diagnostic. L'enfant prélève du sang pour analyse. Cependant, à ce stade, confirmer la présence du syndrome d'Edwards n'est pas le principal problème.
La tâche principale à la naissance d'un enfant atteint de cette pathologie est la détection d'anomalies dans le développement des organes internes, qui entraînent généralement la mort dans les premiers mois de la vie. C'est sur leur recherche que la plupart des procédures de diagnostic sont dirigées immédiatement après la naissance.
Pour détecter les défauts de développement des organes internes, les méthodes de recherche suivantes sont utilisées:
- examen échographique de la cavité abdominale;
- échocardiographie;
- test sanguin général et test sanguin biochimique;
- analyse d'urine générale;
- tomodensitométrie ou imagerie par résonance magnétique ;
- radiographie.
Pour l'examen aux rayons X, la petite enfance est généralement considérée comme une contre-indication, mais dans ce cas, le danger peut être négligé. Le fait est que nous parlons de la détection de pathologies qui présentent un danger pour la vie en ce moment. Par conséquent, les médecins doivent obtenir de toute urgence toutes les informations nécessaires sur les malformations existantes. Cela vous aidera à choisir une stratégie de traitement. Dans la plupart des cas, une bonne prise en charge du patient et des soins appropriés contribuent à prolonger la vie de l'enfant.
Ainsi, nous pouvons conclure qu'il existe de nombreuses méthodes pour diagnostiquer le syndrome d'Edwards. Certains d'entre eux ( amniocentèse, cordocentèse, etc.) représentent un certain risque de complications et ne sont pas réalisées sans indications particulières. Les principales indications sont la présence de cas de maladies chromosomiques dans la famille et l'âge de la mère de plus de 35 ans. Le programme de diagnostic et de prise en charge de la patiente à tous les stades de la grossesse peut être modifié par le médecin traitant si nécessaire.
Pronostic pour les enfants atteints du syndrome d'Edwards
 Compte tenu des multiples troubles du développement inhérents au syndrome d'Edwards, le pronostic des nouveau-nés porteurs de ce diagnostic est presque toujours défavorable. Donnée statistique ( de diverses études indépendantes) disent que plus de la moitié des enfants ( 50 – 55%
) ne vivent pas plus de 3 mois. Moins de dix pour cent des bébés parviennent à fêter leur premier anniversaire. Les enfants qui survivent jusqu'à un âge avancé ont de graves problèmes de santé et ont besoin de soins constants. Pour prolonger la vie, des opérations chirurgicales complexes sur le cœur, les reins ou d'autres organes internes sont souvent nécessaires. La correction des malformations congénitales et des soins qualifiés constants sont, en fait, le seul traitement. Chez les enfants atteints de la forme classique du syndrome d'Edwards ( trisomie complète 18) il n'y a pratiquement aucune chance d'avoir une enfance normale ou une longue vie.
Compte tenu des multiples troubles du développement inhérents au syndrome d'Edwards, le pronostic des nouveau-nés porteurs de ce diagnostic est presque toujours défavorable. Donnée statistique ( de diverses études indépendantes) disent que plus de la moitié des enfants ( 50 – 55%
) ne vivent pas plus de 3 mois. Moins de dix pour cent des bébés parviennent à fêter leur premier anniversaire. Les enfants qui survivent jusqu'à un âge avancé ont de graves problèmes de santé et ont besoin de soins constants. Pour prolonger la vie, des opérations chirurgicales complexes sur le cœur, les reins ou d'autres organes internes sont souvent nécessaires. La correction des malformations congénitales et des soins qualifiés constants sont, en fait, le seul traitement. Chez les enfants atteints de la forme classique du syndrome d'Edwards ( trisomie complète 18) il n'y a pratiquement aucune chance d'avoir une enfance normale ou une longue vie.
Avec une trisomie partielle ou une forme mosaïque du syndrome, le pronostic est un peu meilleur. Dans ce cas, l'espérance de vie moyenne passe à plusieurs années. Cela s'explique par le fait que les anomalies du développement dans les formes les plus bénignes n'entraînent pas aussi rapidement la mort de l'enfant. Néanmoins, le principal problème, à savoir un retard mental grave, est inhérent à tous les patients sans exception. À l'adolescence, il n'y a aucune chance que l'un ou l'autre progéniture continue ( la puberté ne se produit généralement pas), ni la possibilité de travailler ( même mécanique, qui ne nécessite pas de compétences particulières). Il existe des centres spéciaux pour la prise en charge des enfants atteints de maladies congénitales, où les patients atteints du syndrome d'Edwards sont soignés et, si possible, promus. Développement intellectuel. Avec suffisamment d'efforts de la part des médecins et des parents, un enfant qui a vécu plus d'un an peut apprendre à sourire, à réagir au mouvement, à maintenir sa position corporelle ou à manger ( en l'absence de malformations du système digestif). Ainsi, des signes de développement sont encore observés.
La mortalité infantile élevée due à cette maladie s'explique par un grand nombre de malformations des organes internes. Ils sont invisibles directement à la naissance, mais sont présents chez presque tous les patients. Au cours des premiers mois de la vie, les enfants meurent généralement d'un arrêt cardiaque ou respiratoire.
Le plus souvent, des malformations sont observées dans les organes et systèmes suivants :
- système musculo-squelettique ( os et articulations, y compris le crâne);
- le système cardiovasculaire;
- système nerveux central;
- système digestif;
- système urinaire;
- d'autres infractions.
Système musculo-squelettique
Les principales malformations dans le développement du système musculo-squelettique sont la position anormale des doigts et la courbure des pieds. Dans l'articulation de la hanche, les jambes sont rapprochées de telle sorte que les genoux se touchent presque et que les pieds regardent légèrement sur les côtés. Il n'est pas rare que les enfants atteints du syndrome d'Edwards aient un sternum inhabituellement court. Cela déforme la poitrine dans son ensemble et crée des problèmes respiratoires qui s'aggravent avec la croissance, même si les poumons eux-mêmes ne sont pas touchés.
Les malformations du crâne sont principalement cosmétiques. Cependant, des vices tels que la fente palatine, la fente labiale et le palais haut créent de sérieuses difficultés pour nourrir l'enfant. Souvent, avant la chirurgie pour corriger ces défauts, l'enfant est transféré à la nutrition parentérale ( sous forme de compte-gouttes avec des solutions nutritives). Une autre option consiste à utiliser une gastrostomie, un tube spécial par lequel les aliments pénètrent directement dans l'estomac. Sa mise en place nécessite une intervention chirurgicale séparée.
En général, les malformations du système musculo-squelettique ne constituent pas une menace directe pour la vie de l'enfant. Cependant, ils affectent indirectement sa croissance et son développement. La fréquence de tels changements chez les patients atteints du syndrome d'Edwards est d'environ 98%.
Le système cardiovasculaire
Les malformations du système cardiovasculaire sont la principale cause de décès chez les jeunes enfance. Le fait est que de telles violations se produisent dans près de 90% des cas. Le plus souvent, ils perturbent gravement le processus de transport du sang dans l'organisme, entraînant une insuffisance cardiaque grave. La plupart des pathologies cardiaques peuvent être corrigées chirurgicalement, mais tous les enfants ne peuvent pas subir une opération aussi complexe.
Les anomalies les plus courantes du système cardiovasculaire sont :
- non-fermeture du septum interauriculaire ;
- non-fermeture du septum interventriculaire;
- fusion des feuillets valvulaires ( ou, à l'inverse, leur sous-développement);
- coarctation ( constriction) aorte.
Toutes ces malformations cardiaques entraînent de graves troubles circulatoires. Le sang artériel ne coule pas dans le bon volume vers les tissus, c'est pourquoi les cellules du corps commencent à mourir.
système nerveux central
Système digestif
Fréquence des vices système digestif avec le syndrome d'Edwards est jusqu'à 55%. Le plus souvent, ces anomalies du développement constituent une menace sérieuse pour la vie de l'enfant, car elles ne lui permettent pas d'absorber correctement les nutriments. Manger en contournant les organes digestifs naturels affaiblit considérablement le corps et aggrave l'état de l'enfant.
Les malformations les plus courantes du système digestif sont :
- Diverticule de Meckel caecum dans l'intestin grêle);
- atrésie de l'oesophage prolifération de sa lumière, en raison de laquelle la nourriture ne passe pas dans l'estomac);
- atrésie des voies biliaires ( accumulation de bile dans la vessie).
Toutes ces pathologies nécessitent une correction chirurgicale. Dans la plupart des cas, l'opération ne permet que de prolonger légèrement la vie de l'enfant.
système génito-urinaire
Autres infractions
Les autres troubles du développement possibles sont les hernies ( ombilical, inguinal). Des hernies discales de la colonne vertébrale peuvent également être détectées, ce qui entraînera des problèmes neurologiques. Du côté des yeux, on observe parfois une microphtalmie ( petits globes oculaires).
La combinaison de ces malformations prédétermine une mortalité infantile élevée. Dans la plupart des cas, si le syndrome d'Edwards est diagnostiqué au début de la grossesse, les médecins recommanderont un avortement pour des raisons médicales. Cependant, la décision finale est prise par le patient lui-même. Malgré la gravité de la maladie et le mauvais pronostic, de nombreuses personnes préfèrent espérer le meilleur. Mais, malheureusement, dans un avenir proche, des changements majeurs dans les méthodes de diagnostic et de traitement du syndrome d'Edwards ne sont apparemment pas attendus.
- L'essence de la maladie
- Les raisons
- Les symptômes
- Diagnostique
- Traitement
- Prévisions
Après le syndrome de Down, le syndrome d'Edwards est l'anomalie chromosomique la plus fréquente. Le pronostic des enfants atteints d'une telle pathologie étant des plus décevants (issue fatale ou retard mental profond jusqu'à la fin de la vie), les parents ont bien besoin de savoir de quel type de déviation il s'agit, quand peut-on la diagnostiquer afin de faire le bonne décision en temps opportun, qu'il s'agisse de laisser un tel bébé dans l'utérus ou d'interrompre la grossesse.
L'essence de la maladie
Le syndrome de trisomie 18, ou syndrome d'Edwards, est une maladie chromosomique qui se caractérise par un ensemble complexe de malformations multiples et très graves. La raison en est la formation d'un 18e chromosome supplémentaire. Ainsi, le caryotype d'un patient atteint du syndrome d'Edwards est de trois chromosomes 18 au lieu des deux normaux. Dans 90% des cas, il s'agit d'une simple trisomie, moins souvent - mosaïque ou translocation.
La formule de lecture du caryotype du syndrome d'Edwards est indiquée comme suit : 47,XX, 18+ (c'est une fille) et 47,XY, 18+ (c'est un garçon).
à travers les pages de l'histoire. Le syndrome d'Edwards a été décrit pour la première fois par John Edwards en 1960.
Les raisons
La principale cause de la maladie est le chromosome 18 triplé (et non doublé, comme c'est normal) dans le caryotype du zygote. Cependant, ce qui provoque exactement une telle pathologie est encore inconnu des généticiens. De plus, un chromosome supplémentaire apparaît avant même la fécondation. Il y a une opinion parmi les médecins que seuls les faits sont à blâmer, mais le hasard ordinaire. D'autre part, il existe des facteurs de risque qui permettent de séparer dans un groupe spécial les causes présumées du syndrome d'Edwards, qui doivent être prises en compte par les couples sur le point de devenir parents.
- L'âge de la mère est supérieur à 45 ans. Dans ce cas, le risque de syndrome d'Edwards est de 0,7 %.
- Hérédité : si des enfants présentant des anomalies similaires sont déjà nés dans la famille, le risque de donner naissance à un bébé présentant une pathologie du caryotype est très élevé.
- Les mauvaises habitudes restent en vigueur même ici. Certains scientifiques affirment que la forme mosaïque du syndrome d'Edwards (c'est la plus simple) peut être déclenchée par une consommation de drogue à long terme, une énorme quantité de nicotine ou d'alcool dans le corps de l'un des parents.
- Utilisation à long terme de médicaments puissants.
- Infections des voies génitales.
- L'exposition aux rayonnements génère très souvent des mutations chromosomiques.
Ce ne sont là que les causes de l'apparition de trois chromosomes 18 au lieu de deux allégués par les généticiens. Ils ne doivent pas être considérés comme un postulat inébranlable. Ainsi, les facteurs de risque du syndrome d'Edwards sont l'âge de la mère, les maladies chromosomiques déjà présentes dans la famille et les mauvaises habitudes.
Comparée au même syndrome de Down, cette pathologie est un phénomène plutôt rare. De nombreux parents se demandent à quelle fréquence le syndrome d'Edwards est diagnostiqué : selon les statistiques, 1 enfant malade naît sur 7 000 bébés en bonne santé. Étant donné que le pronostic de ces enfants est complètement décevant, il est préférable de se renseigner à l'avance sur le diagnostic.
Fait curieux. C'est encore inexplicable, mais les filles atteintes du syndrome d'Edwards naissent 3 fois plus que les garçons.
Les symptômes
Afin de prendre la bonne décision pendant la grossesse de laisser ou non le bébé avec une telle maladie, les parents doivent clairement comprendre ce que sont les enfants atteints du syndrome d'Edwards à leur naissance. Image clinique la pathologie est sombre et lourde pour le cœur de la mère et du père. Les principaux symptômes peuvent être remarqués dès la naissance du bébé.
À la naissance:
- faible poids (environ 2 kg) et dénutrition prénatale avec durée de grossesse normale voire excessive ;
- asphyxie.
Déformations du visage et du crâne :
- anomalies du crâne, qui a une forme dolichocéphale, un front bas, une nuque saillante;
- ouverture de la bouche et mâchoire inférieure très petites;
- fente palatine et lèvre supérieure;
- strabisme;
- déformation des oreillettes, leur emplacement bas par rapport au visage, allongement dans le plan horizontal;
- absence de lobe et de tragus;
- méat auditif externe rétréci, dans certains cas, il est complètement absent ;
- ptose - relâchement cutané;
- cou court avec un pli cutané caractéristique du col.
Difformités squelettiques :
- sternum court, en raison duquel les espaces intercostaux sont réduits et la poitrine est plus courte et plus large que la normale;
- dans 80% des cas - développement anormal stop : les talons dépassent fortement, les arches s'affaissent (basculement des pieds), les gros orteils sont épaissis et raccourcis ;
- doigts croisés;
- Dislocation congénitale de la hanche;
- pied bot.
Pathologies des organes internes :
- malformations du cœur et des vaisseaux sanguins;
- hypoplasie cérébelleuse;
- pathologie du tractus gastro-intestinal;
- problèmes avec le système génito-urinaire;
- perturbations dans le travail du système nerveux central;
- retard mental sévère.
Développement général :
- difficulté à avaler, sucer, respirer.
Un enfant atteint du syndrome d'Edwards ne pourra jamais vivre pleinement sa vie comme tout le monde. Tout au long de sa courte vie, il surmontera de nombreux obstacles, et il ignorera complètement sa maladie. Le plus difficile est pour les parents qui sont obligés de surveiller l'évolution des pathologies et des malformations avec lesquelles leur bébé est né chaque jour. Quand est-ce que le diagnostic de cette maladie est effectué?
Putain ! Le généticien Edwards, qui a décrit la maladie, a identifié plus de 130 défauts symptomatiques caractéristiques de ce syndrome.
Diagnostique
Le diagnostic du syndrome d'Edwards est très important, car cette pathologie chromosomique est une indication médicale à l'avortement. Les méthodes suivantes sont utilisées pour détecter la maladie.
Échographie et dopplerographie
Il existe des signes particuliers du syndrome d'Edwards à l'échographie que le médecin doit remarquer :
- anomalies multiples dans le développement du fœtus;
- agénésie de l'artère ombilicale;
- petit placenta;
- polyhydramnios.
Mais ce sont tous des signes indirects du syndrome d'Edwards pendant la grossesse, qui doivent être confirmés par des données d'autres études.
Dépistage prénatal standard
Suppose un test sanguin pour la présence de marqueurs sériques :
- de 11 à 13 semaines : βhCG et PAPP ;
- de 20 à 24 semaines : βhCG, estriol libre, α-foetoprotéine.
Si, sur la base de ces tests, la femme enceinte appartient à un groupe à haut risque, des diagnostics supplémentaires lui sont proposés.
Méthodes de diagnostic invasives
Ils sont réalisés dans des cas exceptionnels, lorsque le risque d'avoir un bébé atteint du syndrome d'Edwards est très élevé. Les diagnostics invasifs comprennent :
- biopsie du chorion ;
- amniocentèse;
- cordocentèse;
- caryotype fœtal ultérieur.
Si la maladie n'a pas été détectée à temps et qu'un enfant atteint du syndrome d'Edwards est encore né, il est soumis à un examen complet immédiat.
Diagnostic après la naissance
L'examen d'un enfant malade vise à identifier les malformations. Un nouveau-né atteint du syndrome d'Edwards est examiné par différents spécialistes :
- néonatologiste;
- cardiologue;
- neurologue;
- chirurgien;
- orthopédiste;
- urologue, etc...
Les études diagnostiques les plus importantes réalisées sur un enfant atteint du syndrome d'Edwards immédiatement après sa naissance :
- échocardiographie;
- Échographie des reins;
- Échographie des organes abdominaux.
Certaines caractéristiques du déroulement de la grossesse lors du port d'un enfant atteint du syndrome d'Edwards ne diffèrent pas. Il commence également à bouger en temps voulu, parfois même très actif, aucune toxicose supplémentaire n'est observée. Par conséquent, seuls les équipements de diagnostic modernes peuvent détecter la maladie pendant cette période. Si le bébé est encore né, toute sa courte vie sera consacrée au traitement constant des défauts concomitants.
Avis des médecins. Les filles atteintes du syndrome d'Edwards naissent plus, non pas parce qu'elles sont le plus souvent sensibles à une telle pathologie sur le 18e chromosome. La raison en est leur survie. On pense que la plupart des grossesses de garçons atteints de ce syndrome se terminent par un avortement spontané ou une mort fœtale intra-utérine.
Traitement
Chez ces enfants, les anomalies du développement et toutes sortes de malformations sont incompatibles avec la vie, de sorte que le traitement du syndrome d'Edwards est réduit à des soins symptomatiques. Elle vise à maintenir les fonctions physiologiques de base, à améliorer la qualité de vie et à la prolonger au maximum. La correction chirurgicale des pathologies congénitales, selon la plupart des médecins, est injustifiée et très risquée. Dans les premiers jours de la vie, le traitement est limité à des activités telles que :
- alimentation par sonde en raison de difficultés de déglutition et de succion ;
- IVL prolongé, car il y a des problèmes respiratoires.
Après la sortie (si possible), les parents doivent fournir à un bébé malade :
- soins organisés, presque professionnels : c'est bien s'il y a une personne (nounou) avec une formation médicale à côté de l'enfant ;
- nutrition complète;
- surveillance régulière par divers pédiatres.
Il faut tout de suite faire une réserve que s'occuper d'un enfant atteint du syndrome d'Edwards demande beaucoup de patience et de force.
Prévisions
Les parents dont l'enfant est porteur du syndrome d'Edwards doivent savoir quelles prévisions les attendent. Comme mentionné ci-dessus, ils sont très décevants :
- l'espérance de vie de ces enfants est très faible;
- 60% meurent avant 3 mois (le plus souvent les garçons entrent dans ce groupe) ;
- un autre 10% vivent jusqu'à un an;
- seulement 1 % des personnes nées vivent jusqu'à 10 ans ;
- la cause la plus fréquente de décès est l'insuffisance cardiaque ou l'arrêt respiratoire;
- tous les enfants atteints du syndrome d'Edwards restent profondément oligophréniques jusqu'à la fin de leurs jours ;
- le corps de ces enfants est très affaibli, sujet à toutes sortes de maladies, à cause desquelles ils souffrent souvent d'infections des voies urinaires, de conjonctivite, d'otite moyenne, de sinusite, de pneumonie;
- les compétences de communication verbale chez ces enfants sont très limitées;
- ils peuvent réagir aux paroles de leurs parents ;
- certains sourient même, reconnaissent et interagissent avec les soignants;
- capable d'acquérir des compétences telles que l'auto-alimentation et lever la tête.
Si pendant la grossesse l'enfant a été diagnostiqué avec le syndrome d'Edwards, cela oblige les parents à prendre une décision responsable (le moment du diagnostic de diverses pathologies fœtales peut être trouvé dans l'article suivant). Pourront-ils prendre en charge un enfant gravement malade souffrant de multiples pathologies ? Ou est-il judicieux d'interrompre une telle grossesse? Ici, seul le couple lui-même, par des efforts conjoints, peut trouver la seule bonne issue pour eux.
Pendant la grossesse, les futures mères font un dépistage prénatal pour détecter à l'avance les anomalies génétiques chez le fœtus. L'une des maladies les plus graves de ce groupe est le syndrome décrit par John Edwards en 1960. En médecine, on parle de trisomie 18.
Le syndrome d'Edwards - qu'est-ce que c'est en termes simples ?
Une cellule germinale mâle et femelle saine possède un ensemble standard ou haploïde de chromosomes d'une quantité de 23 pièces. Après avoir fusionné, ils forment un ensemble individuel - un caryotype. C'est comme une sorte de passeport ADN, il contient des données génétiques uniques sur l'enfant. Un caryotype normal ou diploïde contient 46 chromosomes, 2 de chaque espèce, de la mère et du père.
Avec la maladie considérée, il y a un élément dupliqué supplémentaire dans la 18ème paire. C'est la trisomie ou le syndrome d'Edwards - un caryotype composé de 47 chromosomes au lieu de 46 pièces. Parfois, la troisième copie du chromosome 18 est partiellement présente ou ne se trouve pas dans toutes les cellules. De tels cas sont rarement diagnostiqués (environ 5%), ces nuances n'affectent pas l'évolution de la pathologie.
Syndrome d'Edwards - causes
Les généticiens n'ont pas encore compris pourquoi certains enfants ont la mutation chromosomique décrite. Il est considéré comme aléatoire et aucune mesure préventive n'a été développée pour l'empêcher. Certains experts associent des facteurs externes et le syndrome d'Edwards - les raisons qui contribuent vraisemblablement au développement de l'anomalie :
- usage à long terme de drogues, d'alcool, de tabac;
- hérédité;
- l'âge de la mère ou du père est supérieur à 45 ans;
- infections génitales;
- utilisation à long terme de médicaments qui affectent les systèmes immunitaire, endocrinien et système reproducteur;
- exposition aux rayonnements radioactifs.
Syndrome d'Edwards - génétique
Selon des études récentes, le chromosome 18 contient 557 segments d'ADN. Ils codent pour plus de 289 types de protéines dans le corps. À pourcentage c'est 2,5 à 2,6% du matériel génétique, c'est pourquoi le troisième chromosome 18 affecte si fortement le développement du fœtus - le syndrome d'Edwards endommage les os du crâne, les systèmes cardiovasculaire et génito-urinaire. La mutation affecte certaines parties du cerveau et des plexus nerveux périphériques. Un patient atteint du syndrome d'Edwards est caractérisé par un caryotype illustré sur la figure. Cela montre clairement que tous les ensembles sont appariés, à l'exception du 18e ensemble.
Fréquence du syndrome d'Edwards
Cette pathologie est rare, surtout si on la compare à des anomalies génétiques plus connues. La maladie du syndrome d'Edwards est diagnostiquée chez un nouveau-né sur 7 000 bébés en bonne santé, principalement des filles. On ne peut pas affirmer que l'âge du père ou de la mère affecte de manière significative la probabilité de trisomie 18. Le syndrome d'Edwards survient chez les enfants seulement 0,7% plus souvent si les parents ont plus de 45 ans. Cette mutation chromosomique se retrouve également chez les bébés conçus en bas âge.
Syndrome d'Edwards - signes
La maladie considérée a un tableau clinique spécifique qui permet de déterminer avec précision la trisomie 18. Il existe deux groupes de signes qui accompagnent le syndrome d'Edwards - les symptômes sont conditionnellement classés en dysfonctionnement des organes internes et en anomalies externes. Le premier type de manifestation comprend:
- hernie ombilicale, inguinale;
- malformations cardiaques congénitales;
- manque de réflexe de succion et de déglutition;
- diverticule de Meckel ;
- reflux gastro-oesophagien;
- atrésie de l'anus ou de l'œsophage;
- hypertrophie clitoridienne;
- sous-développement du corps calleux, cervelet;
- cryptorchidie;
- emplacement incorrect de l'intestin;
- hypospadias;
- doublement des uretères;
- atrophie ou lissage des circonvolutions cérébrales ;
- rein segmenté ou en fer à cheval;
- ovaires non formés;
- dos incurvé;
- dystrophie musculaire;
- faible poids corporel (environ 2 kg à la naissance).
Extérieurement, il est également facile d'identifier le syndrome d'Edwards - une photo de bébés atteints de trisomie 18 montre la présence des signes suivants :
- petite tête disproportionnée;
- forme de visage déformée;
- fissures palpébrales étroites et courtes;
- oreillettes déformées et basses (allongées horizontalement);
- l'absence de lobe, parfois de tragus et même de conduit auditif ;
- poitrine courte et large;
- mâchoire inférieure sous-développée;
- petite bouche, souvent avec une ouverture triangulaire due à une lèvre supérieure pathologiquement raccourcie;
- pont allongé déprimé du nez;
- "fauteuil à bascule" ;
- sangle entre les doigts ou leur fusion (membres en forme de retournement);
- palais haut, parfois fendu;
- col court avec pli de col proéminent;
- sillons transversaux et coquilles Saint-Jacques sur les paumes ;
- hémangiomes et papillomes sur la peau;
- ptose des paupières;
- strabisme;
- nuque saillante et front bas.
Syndrome d'Edwards - diagnostic
La maladie génétique décrite est une indication directe de l'avortement. Les enfants atteints du syndrome d'Edwards ne pourront jamais vivre pleinement et leur santé se détériorera rapidement. Pour cette raison, il est important de diagnostiquer la trisomie 18 le plus tôt possible. début de mandat. Pour déterminer cette pathologie, plusieurs dépistages informatifs ont été développés.
Analyse pour le syndrome d'Edwards
Il existe des méthodes non invasives et invasives pour étudier le matériel biologique. Le deuxième type de test est considéré comme le plus fiable et fiable, il aide à identifier le syndrome d'Edwards chez le fœtus aux premiers stades de développement. Non invasif est le dépistage prénatal standard du sang de la mère. Les méthodes de diagnostic invasives comprennent :
- Biopsie des villosités choriales. L'étude est menée à partir de 8 semaines. Pour effectuer l'analyse, un morceau de la membrane placentaire est arraché, car sa structure correspond presque parfaitement au tissu du fœtus.
- Amniocentèse. Lors des tests, un échantillon de liquide amniotique est prélevé. Cette procédure détecte le syndrome d'Edwards à partir de 14 semaines de gestation.
- Cordocentèse. L'analyse nécessite un peu de sang de cordon ombilical du fœtus, donc cette méthode de diagnostic n'est utilisée qu'aux stades ultérieurs, à partir de la 20e semaine.
Risque de syndrome d'Edwards par biochimie
Le dépistage prénatal est réalisé au premier trimestre de la grossesse. La future mère doit donner du sang entre 11 et 13 semaines de gestation pour analyse biochimique. Selon les résultats de la détermination du niveau gonadotrophine chorionique et la protéine plasmatique A, le risque de syndrome d'Edwards chez le fœtus est calculé. S'il est élevé, la femme est incluse dans le groupe approprié pour l'étape suivante de la recherche (invasive).
Syndrome d'Edwards - signes à l'échographie
Ce type de diagnostic est rarement utilisé, principalement dans les cas où la femme enceinte n'a pas subi de dépistage génétique préalable. Le syndrome d'Edwards à l'échographie ne peut être détecté qu'aux stades ultérieurs, lorsque le fœtus est déjà presque complètement formé. Symptômes caractéristiques trisomie 18 :
- malformations intra-utérines des systèmes cardiovasculaire et génito-urinaire;
- anomalies des structures musculo-squelettiques;
- pathologie des os du crâne et des tissus mous de la tête.
- Signes indirects de la maladie à l'échographie :
- bradycardie;
- développement fœtal retardé;
- une artère dans le cordon ombilical (devrait être deux );
- hernie dans la cavité abdominale;
- absence visuelle d'os nasaux.
Syndrome d'Edwards - traitement
La thérapie de la mutation considérée vise à soulager ses symptômes et à faciliter la vie du bébé. Il est impossible de guérir le syndrome d'Edwards et d'assurer le plein développement de l'enfant. Les mesures médicales standard aident :
- rétablir le passage des aliments avec atrésie de l'anus ou des intestins ;
- organiser l'alimentation à travers une sonde dans le contexte de l'absence de réflexes de succion et de déglutition;
- stabiliser le fonctionnement du système cardiovasculaire;
- normaliser l'écoulement de l'urine.
Souvent, le syndrome d'Edwards chez les nouveau-nés nécessite en outre l'utilisation de médicaments anti-inflammatoires, antibactériens, hormonaux et autres médicaments puissants. Cela est nécessaire pour une thérapie intensive rapide de toutes les maladies concomitantes qu'elle provoque:
- tumeur de Wilms ;
- conjonctivite;
- pneumonie;
- otite moyenne;
- hypertension pulmonaire;
- sinusite;
- infections urinaires;
- frontite;
- hypertension artérielle et plus encore.
Syndrome d'Edwards - pronostic
La plupart des embryons présentant l'anomalie génétique décrite meurent même pendant la gestation en raison du rejet d'un fœtus inférieur par l'organisme. Après la naissance, le pronostic est également décevant. Si le syndrome d'Edwards est diagnostiqué, combien de temps ces enfants vivent, considérez en pourcentage:
- 60% - pas plus de 3 mois ;
- 7-10 % - 1 an ;
- environ 1% - jusqu'à 10 ans.
Dans des cas exceptionnels (trisomie 18 partielle ou mosaïque), les unités peuvent atteindre la maturité. Même dans de telles situations, le syndrome de John Edwards progressera inexorablement. Les enfants adultes atteints de cette pathologie restent à jamais oligophrènes. Le maximum qu'on puisse leur enseigner :
- lève la tête;
- sourir;
- manger seul;
- reconnaître un cercle restreint de personnes.
Syndrome d'Edwards
trisomie 18
est une maladie congénitale grave causée par des anomalies chromosomiques. C'est l'une des pathologies les plus courantes de cette catégorie (
deuxième seulement après le syndrome de Down en fréquence
). La maladie se caractérise par de nombreux troubles du développement de divers organes et systèmes. Le pronostic d'un enfant est généralement défavorable, mais beaucoup dépend des soins que les parents sont en mesure de lui prodiguer.
La prévalence du syndrome d'Edwards dans le monde varie de 0,015 à 0,02 %. Il n'y a pas de dépendance claire à l'égard de la localité ou de la race. Statistiquement, les filles tombent malades 3 à 4 fois plus souvent que les garçons. Une explication scientifique de cette proportion n'a pas encore été identifiée. Cependant, un certain nombre de facteurs ont été notés qui peuvent augmenter le risque de cette pathologie.
Comme d'autres mutations chromosomiques, le syndrome d'Edwards est, en principe, une maladie incurable. Les méthodes de traitement et de soins les plus modernes ne peuvent que maintenir l'enfant en vie et contribuer à certains progrès de son développement. Il n'y a pas de recommandations uniformes pour la prise en charge de ces enfants en raison de la grande variété de troubles et de complications possibles.
Faits intéressants
- La description des principaux symptômes de cette maladie a été faite au début du XXe siècle.
- Jusqu'au milieu des années 1900, il n'était pas possible de collecter suffisamment d'informations sur cette pathologie. Premièrement, cela nécessitait un niveau de développement technologique approprié qui permettrait la détection d'un chromosome supplémentaire. Deuxièmement, la plupart des enfants meurent dans les premiers jours ou les premières semaines de leur vie en raison du faible niveau de soins médicaux.
- La première description complète de la maladie et de sa cause principale (l'apparition d'un 18e chromosome supplémentaire) n'a été faite qu'en 1960 par le médecin John Edward, d'après qui la nouvelle pathologie a alors été nommée.
- La fréquence réelle du syndrome d'Edwards est de 1 cas pour 2,5 à 3 000 conceptions (0,03 à 0,04%), mais les données officielles sont bien inférieures. Cela est dû au fait que près de la moitié des embryons présentant cette anomalie ne survivent pas et que la grossesse se termine par un avortement spontané ou une mort intra-utérine du fœtus. Un diagnostic détaillé de la cause d'une fausse couche est rarement effectué.
- La trisomie est une variante d'une mutation chromosomique dans laquelle les cellules d'une personne ne contiennent pas 46, mais 47 chromosomes. Il n'y a que 3 syndromes dans ce groupe de maladies. En plus du syndrome d'Edwards, il s'agit du syndrome de Down (trisomie 21 chromosomes) et du syndrome de Patau (trisomie 13 chromosomes). En présence d'autres chromosomes supplémentaires, la pathologie est incompatible avec la vie. Ce n'est que dans ces trois cas que la naissance d'un enfant vivant et sa croissance et son développement ultérieurs (quoique lents) sont possibles.
Causes de la pathologie génétique Le syndrome d'Edwards est maladie génétique qui se caractérise par la présence d'un chromosome supplémentaire dans le génome humain. Pour comprendre les raisons qui provoquent les manifestations visibles de cette pathologie, il est nécessaire de savoir ce que sont les chromosomes eux-mêmes et le matériel génétique dans son ensemble.
Chaque cellule humaine possède un noyau, responsable du stockage et du traitement de l'information génétique. Le noyau contient 46 chromosomes (
), qui sont une molécule à emballage multiple
ADNacide désoxyribonucléique
). Cette molécule contient certaines sections appelées gènes. Chaque gène est le prototype d'un
dans le corps humain. Si nécessaire, la cellule lit les informations de ce prototype et produit la protéine appropriée. Les défauts génétiques conduisent à la production de protéines anormales, qui sont responsables de l'apparition de maladies génétiques.
Une paire de chromosomes est constituée de deux molécules d'ADN identiques (
l'un est paternel, l'autre est maternel
), qui sont reliés entre eux par un petit pont (
centromère
). Le lieu d'adhésion de deux chromosomes dans une paire détermine la forme de l'ensemble de la connexion et son apparence au microscope.
Tous les chromosomes stockent différentes informations génétiques (sur différentes protéines) et sont divisés en groupes suivants :
- groupe A comprend 1 à 3 paires de chromosomes, qui sont grands et en forme de X ;
- groupe B comprend 4 à 5 paires de chromosomes, qui sont également grands, mais le centromère est plus éloigné du centre, c'est pourquoi la forme ressemble à la lettre X avec le centre décalé vers le bas ou vers le haut ;
- groupe C comprend 6 à 12 paires de chromosomes, qui ressemblent aux chromosomes du groupe B en forme, mais leur sont inférieurs en taille;
- groupe D comprend 13 à 15 paires de chromosomes, qui se caractérisent par une taille moyenne et l'emplacement du centromère à la toute fin des molécules, ce qui donne une ressemblance avec la lettre V;
- groupe E comprend 16 à 18 paires de chromosomes, qui se caractérisent par une petite taille et l'emplacement médian du centromère (la forme de la lettre X);
- groupe F comprend 19 à 20 paires de chromosomes, qui sont un peu plus petites que les chromosomes du groupe E et de forme similaire ;
- groupe G comprend 21 à 22 paires de chromosomes, caractérisés par une forme en V et de très petites tailles.
Les 22 paires de chromosomes ci-dessus sont appelées somatiques ou autosomes. De plus, il existe des chromosomes sexuels, qui constituent la 23e paire. Ils ne sont pas similaires en apparence, donc chacun d'eux est désigné séparément. Le chromosome sexuel féminin est désigné X et est similaire au groupe C. Le chromosome sexuel masculin est désigné Y et est similaire en forme et en taille au groupe G. Si l'enfant a les deux chromosomes féminins (type XX), alors une fille est née. Si l'un des chromosomes sexuels est féminin et l'autre masculin, alors un garçon est né (type XY). La formule chromosomique est appelée caryotype et peut être désignée comme suit - 46,XX. Ici, le nombre 46 indique le nombre total de chromosomes (23 paires), et XX est la formule des chromosomes sexuels, qui dépend du sexe (l'exemple montre le caryotype d'une femme normale).
Le syndrome d'Edwards fait référence aux maladies dites chromosomiques, lorsque le problème n'est pas un défaut génétique, mais un défaut de la molécule d'ADN entière. Pour être plus précis, la forme classique de cette maladie implique la présence d'un 18e chromosome supplémentaire. Le caryotype dans de tels cas est désigné par 47,XX, 18+ (
pour filles
) et 47,XY, 18+ (
pour garçon
). Le dernier chiffre indique le numéro du chromosome supplémentaire. Un excès d'informations génétiques dans les cellules conduit à l'apparition des manifestations correspondantes de la maladie, qui sont regroupées sous le nom de "syndrome d'Edwards". La présence d'un complément
) le chromosome numéro 18 a donné un autre (
plus scientifique
) le nom de la maladie est la trisomie 18.
Selon la forme du défaut chromosomique, on distingue trois types de cette maladie:
- Trisomie 18 complète. La forme complète ou classique du syndrome d'Edwards suggère que toutes les cellules du corps ont un chromosome supplémentaire. Cette variante de la maladie survient dans plus de 90% des cas et est la plus sévère.
- Trisomie partielle 18. La trisomie partielle 18 est un phénomène très rare (pas plus de 3 % de tous les cas de syndrome d'Edwards). Avec lui, les cellules du corps ne contiennent pas un chromosome supplémentaire entier, mais seulement un fragment de celui-ci. Un tel défaut peut être le résultat d'une mauvaise division du matériel génétique, mais il est très rare. Parfois, une partie du dix-huitième chromosome est attachée à une autre molécule d'ADN (introduite dans sa structure, allongeant la molécule, ou simplement "s'accroche" avec un pont). La division cellulaire ultérieure conduit au fait que le corps possède 2 chromosomes normaux numéro 18 et une autre partie des gènes de ces chromosomes (un fragment préservé de la molécule d'ADN). Dans ce cas, le nombre de malformations congénitales sera beaucoup plus faible. Il n'y a pas un excès de toute l'information génétique encodée dans le 18e chromosome, mais seulement une partie de celle-ci. Pour les patients atteints de trisomie 18 partielle, le pronostic est meilleur que pour les enfants avec la forme complète, mais reste toujours défavorable.
- forme de mosaïque. La forme mosaïque du syndrome d'Edwards survient dans 5 à 7% des cas de cette maladie. Le mécanisme de son apparition diffère des autres espèces. Le fait est qu'ici le défaut s'est formé après la fusion du spermatozoïde et de l'ovule. Les deux gamètes (cellules sexuelles) avaient initialement un caryotype normal et portaient un chromosome de chaque espèce. Après la fusion, une cellule de formule normale 46,XX ou 46,XY s'est formée. Lors du processus de division de cette cellule, une panne s'est produite. Lors du doublement du matériel génétique, l'un des fragments a reçu un 18e chromosome supplémentaire. Ainsi, à un certain stade, un embryon s'est formé, dont certaines cellules ont un caryotype normal (par exemple, 46, XX) et certaines ont un caryotype du syndrome d'Edwards (47, XX, 18+). La proportion de cellules pathologiques ne dépasse jamais 50 %. Leur nombre dépend du stade de division de la cellule initiale où la panne s'est produite. Plus cela se produit tard, plus la proportion de cellules défectueuses sera faible. La forme tire son nom du fait que toutes les cellules du corps forment une sorte de mosaïque. Certains d'entre eux sont en bonne santé et certains ont une pathologie génétique grave. Dans le même temps, il n'y a pas de modèles dans la distribution des cellules dans le corps, c'est-à-dire que toutes les cellules défectueuses ne peuvent pas être localisées à un seul endroit pour pouvoir être éliminées. L'état général du patient est plus facile qu'avec la forme classique de trisomie 18.
La présence d'un chromosome supplémentaire dans le génome humain pose de nombreux problèmes. Le fait est que les cellules humaines sont programmées pour lire l'information génétique et ne dupliquer que le nombre de molécules d'ADN donné par la nature. Des violations, même dans la structure d'un gène, peuvent entraîner des maladies graves. En présence d'une molécule d'ADN entière, de multiples troubles se développent même au stade du développement intra-utérin avant la naissance d'un enfant.
Selon des études récentes, le chromosome numéro 18 contient 557 gènes qui codent pour au moins 289 protéines différentes. En termes de pourcentage, cela représente environ 2,5 % du matériel génétique total. Les perturbations qu'un tel déséquilibre provoque sont très graves. Une quantité incorrecte de protéines prédétermine de nombreuses anomalies dans le développement de divers organes et tissus. Dans le cas du syndrome d'Edwards, les os du crâne, certaines parties du système nerveux, les systèmes cardiovasculaire et génito-urinaire souffrent plus souvent que d'autres. Apparemment, cela est dû au fait que les gènes situés sur ce chromosome sont liés au développement de ces organes et systèmes.
Ainsi, la principale et unique cause du syndrome d'Edwards est la présence d'une molécule d'ADN supplémentaire. le plus souvent (
dans la forme classique de la maladie
) est hérité de l'un des parents. Normalement, chaque gamète (
sperme et ovule
) contiennent 22 chromosomes somatiques non appariés, plus un chromosome sexuel. Une femme envoie toujours à un enfant un ensemble standard de 22+X, et un homme peut envoyer 22+X ou 22+Y. Cela détermine le sexe de l'enfant. Les cellules germinales des parents sont formées à la suite de la division de cellules ordinaires en deux ensembles. Normalement, la cellule mère se divise en deux parties égales, mais parfois tous les chromosomes ne se divisent pas en deux. Si la 18e paire ne s'est pas dispersée le long des pôles de la cellule, alors l'un des œufs (
ou l'un des spermatozoïdes
) sera défectueux à l'avance. Il n'aura pas 23, mais 24 chromosomes. Si c'est cette cellule qui participe à la fécondation, l'enfant recevra un 18e chromosome supplémentaire.
Les facteurs suivants peuvent affecter une mauvaise division cellulaire :
- Âge des parents. Il a été prouvé que la probabilité d'anomalies chromosomiques augmente en proportion directe avec l'âge de la mère. Dans le syndrome d'Edwards, cette relation est moins prononcée que dans d'autres pathologies similaires (par exemple, le syndrome de Down). Mais pour les femmes de plus de 40 ans, le risque d'avoir un enfant atteint de cette pathologie est en moyenne 6 à 7 fois plus élevé. Une dépendance similaire à l'âge du père est observée dans une bien moindre mesure.
- Tabagisme et alcool. De mauvaises habitudes telles que le tabagisme et l'abus d'alcool peuvent affecter le système reproducteur humain, affectant la division des cellules germinales. Ainsi, l'usage régulier de ces substances (ainsi que d'autres drogues) augmente le risque de mauvaise distribution du matériel génétique.
- Prendre des médicaments. Certains médicaments, s'ils sont mal pris au cours du premier trimestre, peuvent affecter la division des cellules germinales et provoquer une forme mosaïque du syndrome d'Edwards.
- Maladies de la région génitale. Les infections passées avec des dommages aux organes reproducteurs peuvent affecter la division correcte des cellules. Ils augmentent le risque de troubles chromosomiques et génétiques en général, bien que de telles études n'aient pas été menées spécifiquement pour le syndrome d'Edwards.
- rayonnement rayonnement. L'exposition des organes génitaux aux rayons X ou à d'autres rayonnements ionisants peut provoquer des mutations génétiques. Une telle influence externe est particulièrement dangereuse à l'adolescence, lorsque la division cellulaire est la plus active. Les particules qui forment le rayonnement pénètrent facilement dans les tissus et exposent la molécule d'ADN à une sorte de « bombardement ». Si cela se produit au moment de la division cellulaire, le risque de mutation chromosomique est particulièrement élevé.
De manière générale, on ne peut pas dire que les causes du développement du syndrome d'Edwards soient enfin connues et bien étudiées. Les facteurs ci-dessus ne font qu'augmenter le risque de développer cette mutation. La prédisposition congénitale de certaines personnes à une distribution incorrecte du matériel génétique dans les cellules germinales n'est pas exclue. Par exemple, on pense que pour un couple marié qui a déjà donné naissance à un enfant atteint du syndrome d'Edwards, la probabilité d'avoir un deuxième enfant avec une pathologie similaire est de 2 à 3% (environ 200 fois plus élevée que la moyenne prévalence de cette maladie).
À quoi ressemblent les nouveau-nés atteints du syndrome d'Edwards ?
Comme vous le savez, le syndrome d'Edwards peut être diagnostiqué avant la naissance, mais dans la plupart des cas, cette maladie est détectée immédiatement après la naissance d'un enfant. Les nouveau-nés atteints de cette pathologie présentent un certain nombre d'anomalies de développement prononcées, qui permettent parfois de suspecter immédiatement le diagnostic correct. La confirmation est effectuée plus tard à l'aide d'une analyse génétique spéciale.
Les nouveau-nés atteints du syndrome d'Edwards présentent les anomalies développementales caractéristiques suivantes :
- changement de forme du crâne;
- changement de forme des oreilles;
- anomalies dans le développement du ciel;
- chaise berçante pour les pieds;
- longueur anormale des doigts;
- changement de forme de la mâchoire inférieure;
- fusion des doigts;
- anomalies dans le développement des organes génitaux;
- position fléchisseur des mains;
- caractéristiques dermatoglyphiques.
Modification de la forme du crâne Un symptôme typique du syndrome d'Edwards est la dolichocéphalie. C'est le nom d'un changement caractéristique de la forme de la tête d'un nouveau-né, qui se produit également dans certaines autres maladies génétiques. Les dolichocéphales (enfants présentant ce symptôme) ont un crâne plus long et plus étroit. La présence de cette anomalie est précisément confirmée par des mesures particulières. Le rapport de la largeur du crâne au niveau des os pariétaux à la longueur du crâne (de la projection au-dessus de l'arête du nez à l'occiput) est déterminé. Si le rapport résultant est inférieur à 75%, alors cet enfant appartient aux dolichocéphales. En soi, ce symptôme n'est pas une violation grave. Ce n'est qu'un type de forme de crâne que l'on retrouve également chez les personnes tout à fait normales. Les enfants atteints du syndrome d'Edwards dans 80 à 85% des cas sont dolichocéphales prononcés, dans lesquels la disproportion dans la longueur et la largeur du crâne peut être observée même sans mesures spéciales.
Une autre variante d'une anomalie dans le développement du crâne est la soi-disant microcéphalie, dans laquelle la taille de la tête dans son ensemble est trop petite par rapport au reste du corps. Tout d'abord, cela ne s'applique pas au crâne facial (
mâchoires, pommettes, orbites
), à savoir le crâne, dans lequel se trouve le cerveau. La microcéphalie est moins fréquente dans le syndrome d'Edwards que la dolichocéphalie, mais elle survient également à une fréquence plus élevée que chez les personnes en bonne santé.
Changer la forme de l'oreille
Si la dolichocéphalie peut être une variante de la norme, la pathologie du développement de l'oreillette chez les enfants atteints du syndrome d'Edwards est beaucoup plus grave. Dans une certaine mesure, ce symptôme est observé chez plus de 95% des enfants atteints de la forme complète de cette maladie. Avec une forme de mosaïque, sa fréquence est un peu moindre. L'oreillette est généralement située plus bas que chez les personnes normales (
parfois sous le niveau des yeux
). Les renflements caractéristiques du cartilage qui forme l'oreillette sont mal définis ou absents. Le lobe de l'oreille ou le tragus peuvent également être absents (
une petite zone saillante de cartilage devant le conduit auditif
). Le conduit auditif lui-même est généralement rétréci et dans environ 20 à 25%, il est complètement absent.
Anomalies dans le développement du palais Les processus palatins de la mâchoire supérieure se développent ensemble au cours du développement de l'embryon, formant un palais dur. Chez les enfants atteints du syndrome d'Edwards, ce processus reste souvent incomplet. À l'endroit où se trouve la suture médiane chez les personnes normales (elle peut être ressentie au milieu du palais dur avec la langue), elles ont une fente longitudinale.
Il existe plusieurs variantes de ce défaut :
- non-fermeture du palais mou (l'arrière, partie profonde du palais qui surplombe le pharynx) ;
- non-fermeture partielle du palais dur (l'écart ne s'étend pas sur toute la mâchoire supérieure);
- non-fermeture complète du palais dur et mou;
- non-fermeture complète du palais et des lèvres.
Dans certains cas, le dédoublement du ciel est bilatéral. Deux coins saillants de la lèvre supérieure sont le début de fissures pathologiques. L'enfant ne peut pas fermer complètement la bouche à cause de ce défaut. Dans les cas graves, la communication des cavités buccale et nasale est clairement visible (même avec la bouche fermée). Les dents antérieures peuvent manquer ou pousser sur le côté à l'avenir.
Ces défauts de développement sont également connus sous le nom de fente palatine, fente palatine et fente labiale. Tous peuvent survenir en dehors du syndrome d'Edwards, cependant, chez les enfants atteints de cette pathologie, leur fréquence est particulièrement élevée (
près de 20 % des nouveau-nés
). Beaucoup plus fréquemment (
jusqu'à 65% des nouveau-nés
) ont une caractéristique différente connue sous le nom de ciel haut ou gothique. Il peut être attribué aux variantes de la norme, car on le trouve également chez les personnes en bonne santé.
La présence d'une fente palatine ou de la lèvre supérieure ne confirme pas encore le syndrome d'Edwards. Cette malformation peut survenir avec une fréquence assez élevée et indépendamment sans troubles concomitants d'autres organes et systèmes. Il existe un certain nombre d'interventions chirurgicales standard pour corriger cette anomalie.
Pied basculant
C'est le nom d'un changement caractéristique du pied, qui survient principalement dans le cadre du syndrome d'Edwards. Sa fréquence dans cette maladie atteint 75 %. Le défaut réside dans la position incorrecte des os du talus, du calcanéum et du scaphoïde. Il appartient à la catégorie des déformations en valgus plat du pied chez l'enfant.
Extérieurement, le pied d'un nouveau-né ressemble à ceci. Le tubercule calcanéen, sur lequel repose l'arrière du pied, fait saillie vers l'arrière. Dans ce cas, le coffre-fort peut être complètement absent. Ceci est facile à voir en regardant le pied de l'intérieur. Normalement, une ligne concave y apparaît, allant du talon à la base du gros orteil. Avec un stop à bascule, cette ligne est absente. Le pied est plat voire convexe. Cela lui donne une ressemblance avec les pieds d'une chaise berçante.
Longueur de doigt anormale
Chez les enfants atteints du syndrome d'Edwards, une proportion anormale de la longueur des orteils peut être observée dans le contexte de modifications de la structure du pied. En particulier, nous parlons du pouce, qui est normalement le plus long. Chez les nouveau-nés atteints de ce syndrome, il est inférieur en longueur au deuxième doigt. Ce défaut ne peut être vu qu'en redressant les doigts et en les examinant attentivement. Avec l'âge, à mesure que l'enfant grandit, cela devient plus visible. Étant donné que le raccourcissement du gros orteil se produit principalement avec le pied basculant, la prévalence de ces symptômes chez les nouveau-nés est à peu près la même.
Chez l'adulte, le raccourcissement du gros orteil n'a pas une telle valeur diagnostique. Un tel défaut peut être une caractéristique individuelle chez une personne en bonne santé ou une conséquence d'autres facteurs (
déformation articulaire, maladie osseuse, port de chaussures mal ajustées
). À cet égard, ce signe doit être considéré comme un symptôme possible uniquement chez les nouveau-nés en présence d'autres anomalies du développement.
Changer la forme de la mâchoire inférieure
Des modifications de la forme de la mâchoire inférieure chez les nouveau-nés surviennent dans près de 70% des cas. Normalement, le menton chez les enfants ne dépasse pas vers l'avant comme chez les adultes, mais chez les patients atteints du syndrome d'Edwards, il est trop rétracté. Cela est dû au sous-développement de la mâchoire inférieure, appelée micrognathie (
microgénie
). Ce symptôme se retrouve également dans d'autres maladies congénitales. Il n'est pas rare de trouver des adultes avec des traits faciaux similaires. En l'absence de pathologies concomitantes, ceci est considéré comme une variante de la norme, bien qu'il entraîne certaines difficultés.
Les nouveau-nés atteints de micrognathie développent généralement rapidement les problèmes suivants :
- incapacité à garder la bouche fermée pendant une longue période (gouttes de salive);
- difficultés d'alimentation;
- développement tardif des dents et leur emplacement incorrect.
L'écart entre la mâchoire inférieure et supérieure peut être supérieur à 1 cm, ce qui est beaucoup compte tenu de la taille de la tête du bébé.
Fusion des doigts
La fusion des doigts, ou scientifiquement syndactylie, se produit chez environ 45 % des nouveau-nés. Le plus souvent, cette anomalie touche les orteils, mais on retrouve également une syndactylie au niveau des mains. Dans les cas bénins, la fusion est formée par un pli cutané comme une courte membrane. Dans les cas plus graves, une fusion avec des ponts de tissu osseux est observée.
La syndactylie survient non seulement dans le syndrome d'Edwards, mais également dans de nombreuses autres maladies chromosomiques. Il y a aussi des cas où cette malformation était la seule, et sinon le patient ne différait en rien des enfants normaux. À cet égard, la fusion des doigts n'est qu'un des signes possibles du syndrome d'Edwards, ce qui permet de suspecter le diagnostic, mais ne le confirme pas.
Anomalies dans le développement des organes génitaux
Juste après
chez les nouveau-nés atteints du syndrome d'Edwards, des anomalies du développement des organes génitaux externes peuvent parfois être observées. En règle générale, ils sont associés à des défauts de développement de l'ensemble de l'appareil génito-urinaire, mais cela ne peut être établi sans mesures de diagnostic spéciales. Les anomalies les plus fréquentes, visibles de l'extérieur, sont le sous-développement du pénis chez les garçons et l'hypertrophie (
augmenter en taille
) clitoris chez les filles. Ils surviennent dans environ 15 à 20 % des cas. Un peu moins souvent, une localisation anormale de l'urètre peut être observée (
hypospadias
) ou absence de testicules dans le scrotum chez les garçons (
cryptorchidie
Position fléchisseur des mains
La position des fléchisseurs des mains est une disposition spéciale des doigts, causée non pas tant par des troubles structurels dans la région de la main que par une augmentation du tonus musculaire. Les fléchisseurs des doigts et des mains sont constamment tendus, c'est pourquoi le pouce et l'auriculaire semblent recouvrir le reste des doigts, qui sont pressés contre la paume. Ce symptôme est observé dans de nombreuses pathologies congénitales et n'est pas caractéristique du syndrome d'Edwards. Cependant, si une brosse de forme similaire est trouvée, cette pathologie doit être supposée. Avec lui, la position du fléchisseur des doigts est observée chez près de 90% des nouveau-nés.
Caractéristiques dermatoglyphiques De nombreuses anomalies chromosomiques chez les nouveau-nés présentent des modifications dermatoglyphiques caractéristiques (schémas anormaux et plis sur la peau des paumes). Avec le syndrome d'Edwards, certains signes peuvent être retrouvés dans près de 60% des cas. Ils sont importants principalement pour le diagnostic préliminaire en cas de mosaïque ou de forme partielle de la maladie. Avec une trisomie 18 complète, les dermatoglyphes ne sont pas utilisés, car il existe suffisamment d'autres anomalies de développement plus visibles pour suspecter le syndrome d'Edwards.
Les principales caractéristiques dermatoglyphiques du syndrome d'Edwards sont :
- les arcs au bout des doigts sont localisés plus fréquemment que chez les personnes en bonne santé;
- il n'y a pas de pli cutané entre la dernière (ongle) et l'avant-dernière (médiane) phalanges des doigts ;
- 30% des nouveau-nés ont un soi-disant sillon transversal dans la paume (ligne de singe, ligne simienne).
Des études spéciales peuvent révéler d'autres écarts par rapport à la norme, mais immédiatement après la naissance, sans la participation de spécialistes étroits, ces changements suffisent aux médecins.
En plus des signes ci-dessus, il existe un certain nombre d'anomalies de développement possibles qui peuvent aider au diagnostic préliminaire du syndrome d'Edwards. Selon certaines données, avec un examen externe détaillé, jusqu'à 50 signes externes peuvent être détectés. La combinaison des symptômes les plus courants présentés ci-dessus indique avec une forte probabilité que l'enfant présente cette pathologie grave. Avec une variante en mosaïque du syndrome d'Edwards, il peut ne pas y avoir plusieurs anomalies, mais la présence d'une seule d'entre elles est une indication pour un test génétique spécial.
À quoi ressemblent les enfants atteints du syndrome d'Edwards ? Les enfants atteints du syndrome d'Edwards développent généralement une variété de comorbidités en vieillissant. Leurs symptômes commencent à apparaître quelques semaines après la naissance. Ces symptômes peuvent être la première manifestation du syndrome, car avec une variante mosaïque, dans de rares cas, la maladie peut passer inaperçue immédiatement après la naissance. Ensuite, le diagnostic de la maladie devient plus compliqué.
La plupart des manifestations extérieures du syndrome observées à la naissance persistent et deviennent plus perceptibles. Nous parlons de la forme du crâne, du pied basculant, de la déformation de l'oreillette, etc. Peu à peu, d'autres manifestations externes commencent à s'ajouter qui ne pouvaient pas être remarquées immédiatement après la naissance. Dans ce cas, nous parlons de signes pouvant apparaître chez les enfants au cours de la première année de vie.
Les enfants atteints du syndrome d'Edwards ont les caractéristiques externes suivantes :
- retard dans le développement physique;
- pied bot;
- tonus musculaire anormal;
- réactions émotionnelles anormales.
Retard dans le développement physique Le retard dans le développement physique s'explique par le faible poids corporel de l'enfant à la naissance (seulement 2000 - 2200 g à un âge gestationnel normal). Un défaut génétique joue également un rôle important, ce qui ne permet pas à tous les systèmes de l'organisme de se développer normalement et harmonieusement. Les principaux indicateurs permettant d'évaluer la croissance et le développement de l'enfant sont considérablement réduits.
Vous pouvez remarquer l'arriéré d'un enfant par les indicateurs anthropométriques suivants :
- taille de l'enfant ;
- poids de l'enfant;
- Tour de poitrine;
- circonférence de la tête (cet indicateur peut être normal ou même augmenté, mais on ne peut pas s'y fier en raison d'une déformation congénitale du crâne).
Le pied bot est le résultat d'une déformation des os et des articulations des pieds, ainsi que d'un manque de contrôle normal par le système nerveux. Les enfants commencent à peine à marcher (la plupart ne vivent pas à ce stade en raison de malformations congénitales). Extérieurement, la présence d'un pied bot peut être jugée par la déformation des pieds, la position anormale des jambes au repos.
Tonus musculaire anormal
Le tonus anormal, qui à la naissance provoque une position de fléchisseur de la main, commence à se manifester dans d'autres groupes musculaires à mesure qu'il grandit. Le plus souvent, chez les enfants atteints du syndrome d'Edwards, la force musculaire est réduite, ils sont lents et manquent de tonus normal. Selon la nature des dommages au système nerveux central, certains groupes peuvent avoir une augmentation du tonus, qui se manifeste par des contractions spastiques de ces muscles (
ex., fléchisseurs des bras ou extenseurs des jambes
). Extérieurement, cela se manifeste par le manque de coordination minimale des mouvements. Parfois, les contractions spastiques entraînent un entortillement anormal des membres ou même des luxations.
Réactions émotionnelles anormales
L'absence ou la manifestation anormale de toute émotion est le résultat d'anomalies dans le développement de certaines parties du cerveau (
le plus souvent cervelet et corps calleux
). Ces changements entraînent un retard mental grave, qui s'observe chez tous, sans exception, les enfants atteints du syndrome d'Edwards. Extérieurement, un faible niveau de développement se manifeste par une expression faciale "absente" caractéristique, un manque de réponse émotionnelle aux stimuli externes. L'enfant est incapable de maintenir un contact visuel
ne suit pas un doigt se déplaçant devant les yeux, etc.
). L'absence de réponse aux sons aigus peut être le résultat de dommages à la fois au système nerveux et à l'aide auditive. Tous ces signes se retrouvent au fur et à mesure que l'enfant grandit dans les premiers mois de la vie.
À quoi ressemblent les adultes atteints du syndrome d'Edwards ?
Dans la grande majorité des cas, les enfants nés avec le syndrome d'Edwards ne survivent pas jusqu'à l'âge adulte. Dans la forme complète de cette maladie, lorsqu'un chromosome supplémentaire est présent dans chaque cellule du corps, 90 % des enfants meurent avant l'âge de 1 an en raison d'anomalies graves dans le développement des organes internes. Même avec la correction chirurgicale des éventuels défauts et des soins de qualité, leur corps est plus sensible aux maladies infectieuses. Ceci est facilité par les troubles de l'alimentation qui surviennent chez la plupart des enfants. Tout cela explique la mortalité la plus élevée dans le syndrome d'Edwards.
Avec une forme de mosaïque plus douce, lorsque seule une fraction des cellules du corps contient un ensemble anormal de chromosomes, le taux de survie est un peu plus élevé. Cependant, même dans ces cas, seuls quelques patients survivent jusqu'à l'âge adulte. Leur apparence est déterminée par des anomalies congénitales présentes à la naissance (
fente labiale, oreillette déformée, etc.
). Le principal symptôme, présent chez tous les enfants sans exception, est un retard mental grave. Ayant vécu jusqu'à l'âge adulte, un enfant atteint du syndrome d'Edwards est un oligophrène profond (
QI inférieur à 20, ce qui correspond au degré de retard mental le plus sévère
). En général, des cas isolés sont décrits dans la littérature médicale lorsque des enfants atteints du syndrome d'Edwards ont survécu jusqu'à l'âge adulte. De ce fait, trop peu de données objectives ont été accumulées pour parler des signes extérieurs de cette maladie chez l'adulte.
Diagnostic de pathologie génétique
Actuellement, il existe trois étapes principales dans le diagnostic du syndrome d'Edwards, chacune comprenant plusieurs méthodes possibles. Étant donné que cette maladie est incurable, les parents doivent prêter attention aux possibilités de ces méthodes et les utiliser. La plupart des tests sont effectués dans des centres spécialisés pour le diagnostic prénatal, où il y a tout l'équipement nécessaire pour rechercher des maladies génétiques. Cependant, même une consultation avec un généticien ou un néonatologiste peut être utile.
Le diagnostic du syndrome d'Edwards est possible aux stades suivants :
- diagnostic avant la conception;
- diagnostic pendant le développement fœtal;
- diagnostic après la naissance.
Diagnostic avant la conception Le diagnostic avant la conception d'un enfant est une option idéale, mais malheureusement, au stade actuel de développement de la médecine, ses possibilités sont très limitées. Les médecins peuvent utiliser plusieurs méthodes pour suggérer un risque accru d'avoir un enfant atteint d'une anomalie chromosomique, mais pas plus. Le fait est qu'avec le syndrome d'Edwards, en principe, les violations chez les parents ne peuvent pas être détectées. Une cellule sexuelle défectueuse avec 24 chromosomes n'est qu'une parmi plusieurs milliers. Par conséquent, il est impossible de dire avec certitude jusqu'au moment de la conception si un enfant naîtra avec cette maladie.
Les principales méthodes de diagnostic avant la conception sont:
- Histoire de famille. Une histoire familiale est un questionnement détaillé des deux parents sur leur ascendance. Le médecin s'intéresse à tous les cas de maladies héréditaires (et surtout chromosomiques) dans la famille. Si au moins un des parents se souvient d'un cas de trisomie (syndrome d'Edwards, Down, Patau), cela augmente considérablement la probabilité d'avoir un enfant malade. Cependant, le risque est toujours inférieur à 1 %. Avec des cas répétés de ces maladies chez les ancêtres, le risque augmente plusieurs fois. En fait, l'analyse se résume à une consultation avec un néonatologiste ou un généticien. Auparavant, les parents peuvent essayer de collecter des informations plus détaillées sur leurs ancêtres (de préférence 3-4 générations). Cela améliorera la précision de cette méthode.
- Détection des facteurs de risque. Le principal facteur de risque qui augmente objectivement le risque d'anomalies chromosomiques est l'âge de la mère. Comme mentionné ci-dessus, chez les mères après 40 ans, la probabilité d'avoir un enfant atteint du syndrome d'Edwards augmente plusieurs fois. Selon certains rapports, après 45 ans (l'âge de la mère), presque une grossesse sur cinq s'accompagne d'une pathologie chromosomique. La plupart d'entre eux se terminent par une fausse couche. D'autres facteurs sont les maladies infectieuses passées, les maladies chroniques, les mauvaises habitudes. Cependant, leur rôle dans le diagnostic est beaucoup plus faible. Cette méthode ne donne pas non plus de réponse exacte à la question de savoir si un enfant atteint du syndrome d'Edwards sera conçu.
- Analyse génétique des parents. Si les méthodes précédentes se limitaient à interroger les parents, l'analyse génétique est une étude complète qui nécessite un équipement spécial, des réactifs et des spécialistes qualifiés. Le sang est prélevé sur les parents, à partir duquel les leucocytes sont isolés en laboratoire. Après traitement avec des substances spéciales dans ces cellules, les chromosomes au stade de la division deviennent clairement visibles. Ainsi, le caryotype des parents est compilé. Dans la grande majorité des cas, il est normal (avec les troubles chromosomiques que l'on peut retrouver ici, la probabilité de procréation est négligeable). De plus, à l'aide de marqueurs spéciaux (fragments de chaînes moléculaires), il est possible de détecter des sections d'ADN avec des gènes défectueux. Cependant, on ne trouvera pas ici d'anomalies chromosomiques, mais des mutations génétiques qui n'affectent pas directement la probabilité de syndrome d'Edwards. Ainsi, l'analyse génétique des parents avant le moment de la conception, malgré la complexité et le coût élevé, ne donne pas non plus de réponse sans ambiguïté concernant le pronostic de cette pathologie.
Diagnostic au cours du développement fœtal Au cours du développement fœtal, plusieurs moyens permettent de confirmer directement ou indirectement la présence d'une pathologie chromosomique chez l'embryon. La précision de ces méthodes est beaucoup plus élevée, car les médecins ne traitent pas avec les parents, mais avec le fœtus lui-même. L'embryon lui-même et ses cellules avec leur propre ADN sont disponibles pour étude. Cette étape est également appelée diagnostic prénatal et est la plus importante. À ce stade, vous pouvez confirmer le diagnostic, avertir les parents de la présence d'une pathologie et, si nécessaire, interrompre la grossesse. Si la femme décide d'accoucher et que le nouveau-né est vivant, les médecins pourront alors se préparer à l'avance pour lui fournir l'assistance nécessaire.
Les principales méthodes de recherche dans le cadre du diagnostic prénatal sont :
- Examen échographique (échographie). Cette méthode est non invasive, c'est-à-dire qu'elle n'endommage pas les tissus de la mère ou du fœtus. Il est totalement sans danger et est recommandé à toutes les femmes enceintes dans le cadre du diagnostic prénatal (quel que soit leur âge ou leur risque accru de maladies chromosomiques). Le programme standard suppose que l'échographie doit être effectuée trois fois (à 10-14, 20-24 et 32-34 semaines de grossesse). Si le médecin traitant suppose la possibilité de malformations congénitales, une échographie non planifiée peut également être effectuée. Le syndrome d'Edwards peut être indiqué par le retard du fœtus en taille et en poids, une grande quantité de liquide amniotique, des anomalies de développement visibles (microcéphalie, déformation osseuse). Ces troubles sont très susceptibles d'indiquer des maladies génétiques graves, mais le syndrome d'Edwards ne peut pas être définitivement confirmé.
- Amniocentèse. L'amniocentèse est une analyse cytologique (cellulaire) du liquide amniotique. Le médecin insère doucement une aiguille spéciale sous le contrôle d'un échographe. La ponction est faite dans un endroit où il n'y a pas de boucles du cordon ombilical. À l'aide d'une seringue, la quantité de liquide amniotique nécessaire à l'étude est prélevée. La procédure peut être effectuée à tous les trimestres de la grossesse, mais le moment optimal pour le diagnostic des troubles chromosomiques est la période après la 15e semaine de grossesse. La fréquence des complications (jusqu'à l'interruption spontanée de la grossesse) pouvant atteindre 1%, la procédure ne doit donc pas être effectuée en l'absence d'indications. Une fois le liquide amniotique prélevé, le matériau obtenu est traité. Ils contiennent des cellules liquides de la surface de la peau du bébé, qui contiennent des échantillons de son ADN. Ce sont eux qui sont testés pour la présence de maladies génétiques.
- Cordocentèse. La cordocentèse est la méthode la plus informative de diagnostic prénatal. Après anesthésie et sous le contrôle d'un appareil à ultrasons, le médecin perce un vaisseau traversant le cordon ombilical avec une aiguille spéciale. Ainsi, un échantillon de sang (jusqu'à 5 ml) d'un enfant en développement est obtenu. La technique d'analyse est similaire à celle des adultes. Ce matériel peut être examiné avec une grande précision pour diverses anomalies génétiques. Cela inclut le caryotype fœtal. En présence d'un 18e chromosome supplémentaire, on peut parler de syndrome d'Edwards confirmé. Cette analyse est recommandée après la 18ème semaine de grossesse (de manière optimale 22-25 semaines). La fréquence des complications possibles après cordocentèse est de 1,5 à 2%.
- Biopsie chorionique. Le chorion est l'une des membranes germinales contenant des cellules avec l'information génétique du fœtus. Cette étude implique la ponction de l'utérus sous anesthésie à travers la paroi abdominale antérieure. À l'aide d'une pince à biopsie spéciale, un échantillon de tissu est prélevé pour analyse. Ensuite, une étude génétique standard du matériel obtenu est effectuée. Le caryotypage est effectué pour diagnostiquer le syndrome d'Edwards. Le moment optimal pour une biopsie du chorion est considéré comme 9-12 semaines de grossesse. La fréquence des complications est de 2 à 3 %. Le principal avantage qui le distingue des autres méthodes est la rapidité d'obtention du résultat (après 2-4 jours).
Diagnostic après la naissance Le diagnostic du syndrome d'Edwards après la naissance est le plus simple, le plus rapide et le plus précis. Malheureusement, à ce moment-là, un enfant atteint d'une pathologie génétique grave est déjà né, pour lequel il n'existe aucun traitement efficace à notre époque. Si la maladie n'a pas été détectée au stade du diagnostic prénatal (ou si des études appropriées n'ont pas été réalisées), la suspicion de syndrome d'Edwards apparaît immédiatement après la naissance. L'enfant est généralement à terme ou même post-terme, mais son poids est toujours inférieur à la moyenne. De plus, certaines des malformations congénitales mentionnées ci-dessus attirent l'attention. S'ils sont remarqués, une analyse génétique est effectuée pour confirmer le diagnostic. L'enfant prélève du sang pour analyse. Cependant, à ce stade, confirmer la présence du syndrome d'Edwards n'est pas le principal problème.
La tâche principale à la naissance d'un enfant atteint de cette pathologie est la détection d'anomalies dans le développement des organes internes, qui entraînent généralement la mort dans les premiers mois de la vie. C'est sur leur recherche que la plupart des procédures de diagnostic sont dirigées immédiatement après la naissance.
Pour détecter les défauts de développement des organes internes, les méthodes de recherche suivantes sont utilisées:
- examen échographique de la cavité abdominale;
- échocardiographie;
- test sanguin général et test sanguin biochimique;
- analyse d'urine générale;
- tomodensitométrie ou imagerie par résonance magnétique ;
- radiographie.
Pour l'examen aux rayons X, la petite enfance est généralement considérée comme une contre-indication, mais dans ce cas, le danger peut être négligé. Le fait est que nous parlons de la détection de pathologies qui présentent un danger pour la vie en ce moment. Par conséquent, les médecins doivent obtenir de toute urgence toutes les informations nécessaires sur les malformations existantes. Cela vous aidera à choisir une stratégie de traitement. Dans la plupart des cas, une bonne prise en charge du patient et des soins appropriés contribuent à prolonger la vie de l'enfant.
Ainsi, nous pouvons conclure qu'il existe de nombreuses méthodes pour diagnostiquer le syndrome d'Edwards. Certains d'entre eux (
amniocentèse, cordocentèse, etc.
) représentent un certain risque de complications et ne sont pas réalisées sans indications particulières. Les principales indications sont la présence de cas de maladies chromosomiques dans la famille et l'âge de la mère de plus de 35 ans. Le programme de diagnostic et de prise en charge de la patiente à tous les stades de la grossesse peut être modifié par le médecin traitant si nécessaire.
Pronostic pour les enfants atteints du syndrome d'Edwards
Compte tenu des multiples troubles du développement inhérents au syndrome d'Edwards, le pronostic des nouveau-nés porteurs de ce diagnostic est presque toujours défavorable. Donnée statistique (
de diverses études indépendantes
) disent que plus de la moitié des enfants (
) ne vivent pas plus de 3 mois. Moins de dix pour cent des bébés parviennent à fêter leur premier anniversaire. Les enfants qui survivent jusqu'à un âge avancé ont de graves problèmes de santé et ont besoin de soins constants. Des interventions chirurgicales compliquées sont souvent nécessaires pour prolonger la vie.
Reins ou autres organes internes. La correction des malformations congénitales et des soins qualifiés constants sont, en fait, le seul traitement. Chez les enfants atteints de la forme classique du syndrome d'Edwards (
trisomie complète 18
) il n'y a pratiquement aucune chance d'avoir une enfance normale ou une longue vie.
Avec une trisomie partielle ou une forme mosaïque du syndrome, le pronostic est un peu meilleur. Dans ce cas, l'espérance de vie moyenne passe à plusieurs années. Cela s'explique par le fait que les anomalies du développement dans les formes les plus bénignes n'entraînent pas aussi rapidement la mort de l'enfant. Néanmoins, le principal problème, à savoir un retard mental grave, est inhérent à tous les patients sans exception. À l'adolescence, il n'y a aucune chance que l'un ou l'autre progéniture continue (
la puberté ne se produit généralement pas
), ni la possibilité de travailler (
même mécanique, qui ne nécessite pas de compétences particulières
). Il existe des centres spéciaux pour la prise en charge des enfants atteints de maladies congénitales, où les patients atteints du syndrome d'Edwards sont soignés et, si possible, favorisent leur développement intellectuel. Avec suffisamment d'efforts de la part des médecins et des parents, un enfant qui a vécu plus d'un an peut apprendre à sourire, à réagir au mouvement, à maintenir sa position corporelle ou à manger (
en l'absence de malformations du système digestif
). Ainsi, des signes de développement sont encore observés.
La mortalité infantile élevée due à cette maladie s'explique par un grand nombre de malformations des organes internes. Ils sont invisibles directement à la naissance, mais sont présents chez presque tous les patients. Au cours des premiers mois de la vie, les enfants meurent généralement d'un arrêt cardiaque ou respiratoire.
Le plus souvent, des malformations sont observées dans les organes et systèmes suivants :
- système musculo-squelettique (os et articulations, y compris le crâne);
- le système cardiovasculaire;
- système nerveux central;
- système digestif;
- système urinaire;
- d'autres infractions.
Système musculo-squelettique Les principales malformations dans le développement du système musculo-squelettique sont la position anormale des doigts et la courbure des pieds. Dans l'articulation de la hanche, les jambes sont rapprochées de telle sorte que les genoux se touchent presque et que les pieds regardent légèrement sur les côtés. Il n'est pas rare que les enfants atteints du syndrome d'Edwards aient un sternum inhabituellement court. Cela déforme la poitrine dans son ensemble et crée des problèmes respiratoires qui s'aggravent avec la croissance, même si les poumons eux-mêmes ne sont pas touchés.
Les malformations du crâne sont principalement cosmétiques. Cependant, des vices tels que la fente palatine, la fente labiale et le palais haut créent de sérieuses difficultés pour nourrir l'enfant. Souvent, avant la chirurgie pour corriger ces défauts, l'enfant est transféré à la nutrition parentérale (
sous forme de compte-gouttes avec des solutions nutritives
). Une autre option consiste à utiliser une gastrostomie, un tube spécial par lequel les aliments pénètrent directement dans l'estomac. Sa mise en place nécessite une intervention chirurgicale séparée.
En général, les malformations du système musculo-squelettique ne constituent pas une menace directe pour la vie de l'enfant. Cependant, ils affectent indirectement sa croissance et son développement. La fréquence de tels changements chez les patients atteints du syndrome d'Edwards est d'environ 98%.
Le système cardiovasculaire
Les malformations du système cardiovasculaire sont la première cause de décès dans la petite enfance. Le fait est que de telles violations se produisent dans près de 90% des cas. Le plus souvent, ils perturbent gravement le processus de transport du sang dans l'organisme, entraînant de graves
insuffisance cardiaque
La plupart des pathologies cardiaques peuvent être corrigées chirurgicalement, mais tous les enfants ne peuvent pas subir une opération aussi complexe.
Les anomalies les plus courantes du système cardiovasculaire sont :
- non-fermeture du septum interauriculaire ;
- non-fermeture du septum interventriculaire;
- fusion des feuillets valvulaires (ou, au contraire, leur sous-développement);
- coarctation (rétrécissement) de l'aorte.
Toutes ces malformations cardiaques entraînent de graves troubles circulatoires. Le sang artériel ne coule pas dans le bon volume vers les tissus, c'est pourquoi les cellules du corps commencent à mourir.
système nerveux central
Le défaut le plus caractéristique du côté du système nerveux central est le sous-développement du corps calleux et du cervelet. C'est la cause d'une grande variété de troubles, dont le retard mental, qui est observé chez 100% des enfants. De plus, des troubles au niveau du cerveau et de la moelle épinière provoquent un tonus musculaire anormal et une prédisposition à
convulsions
ou des contractions musculaires spastiques.
Système digestif
La fréquence des malformations du système digestif dans le syndrome d'Edwards peut atteindre 55%. Le plus souvent, ces anomalies du développement constituent une menace sérieuse pour la vie de l'enfant, car elles ne lui permettent pas d'absorber correctement les nutriments. Manger en contournant les organes digestifs naturels affaiblit considérablement le corps et aggrave l'état de l'enfant.
Les malformations les plus courantes du système digestif sont :
- Diverticule de Meckel (caecum dans l'intestin grêle);
- atrésie de l'œsophage (excroissance de sa lumière, à cause de laquelle la nourriture ne passe pas dans l'estomac);
- atrésie des voies biliaires (accumulation de bile dans la vessie).
Toutes ces pathologies nécessitent une correction chirurgicale. Dans la plupart des cas, l'opération ne permet que de prolonger légèrement la vie de l'enfant.
système génito-urinaire
Les malformations les plus graves du système génito-urinaire sont associées à une violation des reins. Dans certains cas, une atrésie des uretères est observée. Le rein d'un côté peut être dupliqué ou fusionné avec des tissus adjacents. En cas de violation de la filtration, les déchets toxiques commencent à s'accumuler dans le corps au fil du temps. De plus, il peut y avoir une augmentation
pression artérielle
et des perturbations dans le travail du cœur. De graves anomalies dans le développement des reins constituent une menace directe pour la vie.
Autres infractions
D'autres troubles du développement possibles sont
hernie (ombilicale, inguinale)
peut être trouvé et
hernie discale de la colonne vertébrale
Ce qui entraînera des problèmes neurologiques. Du côté des yeux, on observe parfois une microphtalmie (
petits globes oculaires
La combinaison de ces malformations prédétermine une mortalité infantile élevée. Dans la plupart des cas, si le syndrome d'Edwards est diagnostiqué au début de la grossesse, les médecins recommanderont un avortement pour des raisons médicales. Cependant, la décision finale est prise par le patient lui-même. Malgré la gravité de la maladie et le mauvais pronostic, de nombreuses personnes préfèrent espérer le meilleur. Mais, malheureusement, dans un avenir proche, des changements majeurs dans les méthodes de diagnostic et de traitement du syndrome d'Edwards ne sont apparemment pas attendus.
Moderne méthodes de diagnostic permettent d'identifier un grand nombre de maladies congénitales au stade de porter un enfant. Le syndrome d'Edwards est l'une des pathologies les plus dangereuses entraînant le développement d'un grand nombre de troubles. En médecine, la maladie s'appelle Trisomie 18.
Et maintenant, arrêtons-nous là-dessus plus en détail.
Qu'est-ce que le syndrome d'Edwards ?
Le syndrome d'Edwards est une anomalie chromosomique qui entraîne toute une liste de troubles et d'anomalies dans le développement de l'enfant. Des changements se produisent dans le chromosome 18. Chez les patients atteints du syndrome, ses copies supplémentaires sont observées. Ce fait provoque l'apparition de complications de nature génétique.
L'incidence de la maladie est de 1 cas sur 7000 enfants en bonne santé. La plupart des nouveau-nés nés avec cette déviation meurent dans les premières semaines de vie. Environ 10 % seulement des enfants vivent un an. La maladie entraîne l'apparition d'un retard mental profond, de lésions congénitales des organes internes et externes. Il y a souvent des défauts du cœur, des reins, du cerveau. Les enfants atteints de cette pathologie ont une petite tête et une petite mâchoire. Il y a une fente labiale ou palatine. Le pied bot est présent.
La maladie a été décrite pour la première fois en 1960. Les symptômes de la maladie ont été formés et décrits par D. Edwards. Il a pu établir la relation entre un certain nombre de signes et a trouvé plus de 130 défauts associés à la maladie. Les symptômes de la pathologie sont très prononcés. Cependant, les traitements actuels ne sont pas efficaces contre eux. Il existe trois types de maladie. La liste comprend :
- Trisomie simple. La maladie est détectée dans 90% des cas.
- Trisomie mosaïque. Les chromosomes endommagés ne sont présents que dans certaines cellules.
- Translocation.
Selon les statistiques, les maladies sont détectées chez les filles 3 fois plus souvent que chez les garçons. Le risque qu'un nouveau-né soit atteint du syndrome d'Edwards augmente avec l'âge de la mère. La probabilité d'avoir un tel enfant est la plus élevée si la femme a plus de 30 ans. Après 45 ans, le risque passe à 0,7 %.
La pathologie affecte considérablement l'espérance de vie. Les garçons ne vivent généralement que jusqu'à 3 mois. Les filles malades vivent plus longtemps. Il est à noter que ces bébés ont atteint l'âge de dix mois. Cependant, ces enfants ne passent pratiquement pas au stade de la croissance. Les causes de mort fœtale sont des infections pulmonaires ou une suffocation, un dysfonctionnement du cœur ou des vaisseaux sanguins, ainsi qu'un tractus intestinal cassé ou obstrué. Tous les phénomènes ci-dessus se produisent à la suite d'une pathologie.
À quoi ressemble le syndrome d'Edwards avec une photo
Les méthodes modernes permettent de diagnostiquer la pathologie avant la naissance d'un enfant. Cependant, dans la plupart des cas, la maladie est détectée après la naissance du nouveau-né. Les enfants atteints du syndrome d'Edwards ont des anomalies de développement prononcées. Ils permettent de suspecter immédiatement le bon diagnostic. La confirmation est effectuée à l'aide d'une analyse spéciale.
Les bébés nés avec le syndrome d'Edwards se distinguent facilement des autres par leur apparence. Ces enfants ont :
- doigts épissés;
- bascule du pied
- forme altérée du crâne;
- anomalies dans le développement des organes génitaux;
- changement de forme de la mâchoire inférieure;
- anomalie dans le développement du palais;
- caractéristiques dermatoglyphiques ;
- changement de forme des oreilles;
- position fléchisseur des mains.
Typique manifestation extérieure il y a un changement dans la forme de la tête d'un nouveau-né. Les enfants atteints de ce syndrome ont un crâne plus long et plus étroit. L'anomalie est confirmée par des mesures spéciales. Une autre option pour l'anomalie est la présence d'une tête trop petite par rapport au reste du corps. Ce phénomène affecte précisément le crâne, dans lequel se trouve le cerveau.
Tous les enfants ayant un problème similaire ont des pathologies dans le développement des oreillettes. Ce symptôme survient dans 95% des cas. Cependant, avec la forme mosaïque du syndrome d'Edwards, le risque d'apparition d'une anomalie est réduit. L'oreillette chez ces enfants est située beaucoup plus bas. Parfois, il est en dessous du niveau des yeux. Le renflement caractéristique du cartilage qui forme l'oreillette est absent. Le conduit auditif lui-même est rétréci. Dans 20 à 25 % des cas, il peut être complètement absent.
Chez les enfants, le processus de formation du palais dur au cours du développement fœtal reste incomplet. Là où une suture médiane est observée chez les personnes en bonne santé, un écart longitudinal est présent chez les patients atteints de la maladie.
Il y a un endroit pour être une chaise berçante. Le défaut consiste en un emplacement incorrect du talus, du calcanéus et des os naviculaires. La pathologie appartient à la catégorie des patients atteints de déformation du pied plat chez les enfants. Le pied externe d'un nouveau-né se distingue par l'abduction du dos du tubercule calcanéen. Dans ce cas, la voûte plantaire peut être complètement absente.
Les patients atteints de pathologie se caractérisent par une proportion anormale de longueurs de doigts. Normalement, le pouce devrait être le plus long. Chez les nouveau-nés atteints du syndrome d'Edwards, sa longueur est inférieure à la seconde. Au fur et à mesure que l'enfant grandit, cette caractéristique devient de plus en plus perceptible. L'épissage des doigts est possible.
Il y a un changement dans la forme de la mâchoire inférieure. Chez les patients atteints du syndrome d'Edwards, le menton est fortement rétracté. Cela est dû au sous-développement de la mâchoire inférieure.
Les premiers signes du syndrome d'Edwards
Les premiers signes du développement de la pathologie sont perceptibles même dans le processus de développement intra-utérin. Pendant la grossesse, il y a un petit placenta, ainsi que des polyhydramnios. La mobilité fœtale est réduite. Une seule artère ombilicale est présente. L'enfant naît avec un poids corporel d'un peu plus de 2 kg. De plus, une malnutrition prénatale peut être présente. Dans certains cas, d'autres signes du syndrome d'Edwards sont également détectés. Ainsi, il peut être détecté avec fixation à la naissance.
Les caractéristiques phénotypiques sont clairement visibles. Les enfants ont un front bas et une petite bouche. Il y a une déformation des oreillettes et un occiput saillant. Il y a une fente palatine et la lèvre supérieure. Il y a ptose et déformation du squelette. Il y a une forme caractéristique du crâne. Parfois, une anomalie des côtes et une luxation congénitale de la hanche se développent. Le tragus peut être présent sur les oreilles. Le bébé a un cou court. Il y a un strabisme et un pied bot. Les reins sont petits. Les doigts croisés sont observés. Le pied a la forme d'un fauteuil à bascule. Les papillomes et sont présents sur la peau.
Symptômes du syndrome d'Edwards
Les principaux symptômes de la maladie sont des changements externes et internes. Ils surviennent à la suite du développement de la pathologie. Ces enfants ont une gueule de loup et une fente labiale. Ils ont une apparence caractéristique, un faible poids à la naissance. De plus, il y a une place pour les anomalies neuropsychiques. La liste des pathologies liées au fonctionnement des organes internes et de la motricité comprend :
- Un problème lié au travail du système nerveux central. De nombreux enfants ont des retards de développement neurologique. Il y a un retard mental. Il y a un sous-développement du cervelet et du corps calleux. Il y a lissage ou atrophie complète des circonvolutions cérébrales.
- Problèmes avec le système cardiovasculaire. La liste des pathologies est assez longue. Ainsi, un foramen ovale ouvert, un canal artériel similaire et un défaut septal ventriculaire peuvent être présents.
- Pathologies du développement de l'appareil génito-urinaire. Les garçons ont la cryptorchidie et. Chez les filles, il y a un sous-développement des ovaires et une hypertrophie du clitoris. Indépendamment du sexe, des changements pathologiques dans les reins ont lieu. Il y a un dédoublement des uretères.
- Maladies du système digestif. Il existe un reflux gastro-oesophagien. Les réflexes de déglutition et de succion sont altérés. Il existe une atrésie de l'œsophage ou de l'anus. Il y a une violation de l'emplacement de l'intestin.
De plus, un strabisme et une scoliose peuvent être présents. Une atrophie musculaire est observée.
Causes et prévention du syndrome d'Edwards
Le syndrome d'Edwards est dû à une non-disjonction myope des chromosomes. Le processus est effectué au stade de la spermatogenèse ou de la gamétogenèse. Dans près de 90% des cas, la maladie est représentée par un simple trypanosome. Moins souvent, des réarrangements déséquilibrés des chromosomes ou une variété de mosaïque de la maladie se produisent. Les causes du syndrome d'Edwards sont généralement dues à l'apparition d'un chromosome maternel supplémentaire dans le code génétique.
Les généticiens n'ont pas encore été en mesure de déterminer pourquoi une telle mutation chromosomique se produit. On pense qu'un tel phénomène est aléatoire et qu'aucun facteur externe ne l'affecte. Cependant, un certain nombre de raisons contribuent à anomalies chromosomiques, révélé aujourd'hui. La liste comprend :
- infections génitales chez les parents;
- hérédité;
- les parents ont plus de 45 ans;
- pendant la grossesse, la mère et le fœtus ont été exposés à des rayonnements radioactifs ;
- une femme utilise depuis longtemps des médicaments qui affectent les systèmes immunitaire, endocrinien et reproducteur (une règle similaire s'applique aux hommes);
- il y a une consommation à long terme de tabac, de drogues et d'alcool.
Comme toutes les pathologies génétiques, le syndrome d'Edwards ne peut être prévenu par méthodes préventives. Pour réduire le risque de maladie, les parents ne peuvent pré-passer que des études de diagnostic qui vous permettent de déterminer à l'avance la probabilité d'une mutation. De plus, pendant la grossesse, la femme et le fœtus doivent être strictement surveillés. La procédure de dépistage prénatal et un certain nombre d'autres études ne peuvent être négligées.
Traitement du syndrome d'Edwards
Il faut garder à l'esprit que seul un enfant sur dix né avec une pathologie similaire vit jusqu'à un an. Par conséquent, le traitement initial consiste à se débarrasser des maladies qui augmentent le risque de décès. En cas d'atrésie intestinale ou anale, des mesures de défécation sont utilisées. L'enfant est alimenté par un tube. Si des processus inflammatoires ou infectieux actifs sont observés dans le corps d'un nouveau-né, un traitement thérapeutique est effectué.
Si l'enfant peut encore être sauvé, la chirurgie peut être effectuée plus tard. Tout d'abord, l'enlèvement de la gueule du loup est effectué. De plus, il y a une lutte contre les malformations cardiaques. Ombilical et retiré à l'aide de méthodes chirurgicales.
Il existe également une thérapie médicamenteuse. On prescrit à l'enfant des médicaments qui peuvent le soulager de la constipation, de l'acidité élevée et des flatulences. Les parents doivent comprendre que le syndrome d'Edwards contribue à l'apparition des pathologies suivantes :
- néoplasme dans les reins;
- hypertension pulmonaire;
- pneumonie;
- hypertension artérielle;
- sinusite et sinusite frontale;
- maladies infectieuses du système génito-urinaire.
En raison du grand nombre de changements pathologiques survenant dans les organes internes et externes du patient, le pronostic de développement ultérieur est défavorable. Les enfants qui parviennent à vivre jusqu'à un an et atteignent un âge relativement adulte auront une nette inhibition mentale du développement. Ils ne pourront pas prendre soin d'eux-mêmes et satisfaire leur exigences naturelles. Pour ces patients, il est nécessaire d'assurer une prise en charge et un suivi constants tout au long de leur vie.
Cependant, les enfants handicapés comprennent quand on communique affectueusement avec eux, qu'on joue avec eux et qu'on les console. Les enfants atteints de ce syndrome peuvent apprendre à manger et à sourire par eux-mêmes. Il est possible d'apprendre progressivement d'autres compétences ménagères utiles.
Un nombre élevé de pathologies des organes internes qui se sont mal développées entraîne un pourcentage élevé de mortalité infantile. Si, au cours des premiers mois de la grossesse, une femme est à risque de contracter cette maladie, les médecins recommandent de pratiquer un avortement pour des raisons médicales. Cependant, la décision finale appartient à la femme.
Après le syndrome de Down, le syndrome d'Edwards est l'anomalie chromosomique la plus fréquente. Le pronostic des enfants atteints d'une telle pathologie étant des plus décevants (issue fatale ou retard mental profond jusqu'à la fin de la vie), les parents ont bien besoin de savoir de quel type de déviation il s'agit, quand peut-on la diagnostiquer afin de faire le bonne décision en temps opportun, qu'il s'agisse de laisser un tel bébé dans l'utérus ou d'interrompre la grossesse.
Le syndrome de trisomie 18, ou syndrome d'Edwards, est une maladie chromosomique qui se caractérise par un ensemble complexe de malformations multiples et très graves. La raison en est la formation d'un 18e chromosome supplémentaire. Ainsi, le caryotype d'un patient atteint du syndrome d'Edwards est de trois chromosomes 18 au lieu des deux normaux. Dans 90% des cas, il s'agit d'une simple trisomie, moins souvent - mosaïque ou translocation.
La formule de lecture du caryotype du syndrome d'Edwards est indiquée comme suit : 47,XX, 18+ (c'est une fille) et 47,XY, 18+ (c'est un garçon).
à travers les pages de l'histoire. Le syndrome d'Edwards a été décrit pour la première fois par John Edwards en 1960.
Les raisons

La principale cause de la maladie est le chromosome 18 triplé (et non doublé, comme c'est normal) dans le caryotype du zygote. Cependant, ce qui provoque exactement une telle pathologie est encore inconnu des généticiens. De plus, un chromosome supplémentaire apparaît avant même la fécondation. Il y a une opinion parmi les médecins que seuls les faits sont à blâmer, mais le hasard ordinaire. D'autre part, il existe des facteurs de risque qui permettent de séparer dans un groupe spécial les causes présumées du syndrome d'Edwards, qui doivent être prises en compte par les couples sur le point de devenir parents.
- L'âge de la mère est supérieur à 45 ans. Dans ce cas, le risque de syndrome d'Edwards est de 0,7 %.
- Hérédité : si des enfants présentant des anomalies similaires sont déjà nés dans la famille, le risque de donner naissance à un bébé présentant une pathologie du caryotype est très élevé.
- Les mauvaises habitudes restent en vigueur même ici. Certains scientifiques affirment que la forme mosaïque du syndrome d'Edwards (c'est la plus simple) peut être déclenchée par une consommation de drogue à long terme, une énorme quantité de nicotine ou d'alcool dans le corps de l'un des parents.
- Utilisation à long terme de médicaments puissants.
- Infections des voies génitales.
- L'exposition aux radiations génère très souvent.
Ce ne sont là que les causes de l'apparition de trois chromosomes 18 au lieu de deux allégués par les généticiens. Ils ne doivent pas être considérés comme un postulat inébranlable. Ainsi, les facteurs de risque du syndrome d'Edwards sont l'âge de la mère, les maladies chromosomiques déjà présentes dans la famille et les mauvaises habitudes.
Comparée à la même, cette pathologie est un événement plutôt rare. De nombreux parents se demandent à quelle fréquence le syndrome d'Edwards est diagnostiqué : selon les statistiques, 1 enfant malade naît sur 7 000 bébés en bonne santé. Étant donné que le pronostic de ces enfants est complètement décevant, il est préférable de se renseigner à l'avance sur le diagnostic.
Fait curieux. C'est encore inexplicable, mais les filles atteintes du syndrome d'Edwards naissent 3 fois plus que les garçons.
Les symptômes

Afin de prendre la bonne décision pendant la grossesse de laisser ou non le bébé avec une telle maladie, les parents doivent clairement comprendre ce que sont les enfants atteints du syndrome d'Edwards à leur naissance. Le tableau clinique de la pathologie est sombre et difficile pour le cœur de la mère et du père. Les principaux symptômes peuvent être remarqués dès la naissance du bébé.
À la naissance:
- faible poids (environ 2 kg) et dénutrition prénatale avec durée de grossesse normale voire excessive ;
- asphyxie.
Déformations du visage et du crâne :
- anomalies du crâne, qui a une forme dolichocéphale, un front bas, une nuque saillante;
- ouverture de la bouche et mâchoire inférieure très petites;
- fente palatine et lèvre supérieure;
- fissures palpébrales étroites et courtes;
- strabisme;
- déformation des oreillettes, leur emplacement bas par rapport au visage, allongement dans le plan horizontal;
- absence de lobe et de tragus;
- méat auditif externe rétréci, dans certains cas, il est complètement absent ;
- ptose - relâchement cutané;
- cou court avec un pli cutané caractéristique du col.
Difformités squelettiques :
- sternum court, en raison duquel les espaces intercostaux sont réduits et la poitrine est plus courte et plus large que la normale;
- dans 80% des cas - développement anormal des pieds : les talons dépassent fortement, les arches s'affaissent (basculement des pieds), les gros orteils sont épaissis et raccourcis ;
- doigts croisés;
- Dislocation congénitale de la hanche;
- pied bot.
Pathologies des organes internes :
- malformations du cœur et des vaisseaux sanguins;
- hypoplasie cérébelleuse;
- pathologie du tractus gastro-intestinal;
- problèmes avec le système génito-urinaire;
- perturbations dans le travail du système nerveux central;
- retard mental sévère.
Développement général :
- difficulté à avaler, sucer, respirer.
Un enfant atteint du syndrome d'Edwards ne pourra jamais vivre pleinement sa vie comme tout le monde. Tout au long de sa courte vie, il surmontera de nombreux obstacles, et il ignorera complètement sa maladie. Le plus difficile est pour les parents qui sont obligés de surveiller l'évolution des pathologies et des malformations avec lesquelles leur bébé est né chaque jour. Quand est-ce que le diagnostic de cette maladie est effectué?
Putain ! Le généticien Edwards, qui a décrit la maladie, a identifié plus de 130 défauts symptomatiques caractéristiques de ce syndrome.
Diagnostique

Le diagnostic du syndrome d'Edwards est très important, car cette pathologie chromosomique est une indication médicale à l'avortement. Les méthodes suivantes sont utilisées pour détecter la maladie.
Échographie et dopplerographie
Il existe des signes particuliers du syndrome d'Edwards à l'échographie que le médecin doit remarquer :
- anomalies multiples dans le développement du fœtus;
- agénésie de l'artère ombilicale;
- petit placenta;
Mais ce sont tous des signes indirects du syndrome d'Edwards pendant la grossesse, qui doivent être confirmés par des données d'autres études.
Dépistage prénatal standard
Suppose un test sanguin pour la présence de marqueurs sériques :
- de 11 à 13 semaines : βhCG et PAPP ;
- de 20 à 24 semaines : βhCG, estriol libre, α-foetoprotéine.
Si, sur la base de ces tests, la femme enceinte appartient à un groupe à haut risque, des diagnostics supplémentaires lui sont proposés.
Méthodes de diagnostic invasives

Ils sont réalisés dans des cas exceptionnels, lorsque le risque d'avoir un bébé atteint du syndrome d'Edwards est très élevé. Les diagnostics invasifs comprennent :
- biopsie du chorion ;
- caryotype fœtal ultérieur.
Si la maladie n'a pas été détectée à temps et qu'un enfant atteint du syndrome d'Edwards est encore né, il est soumis à un examen complet immédiat.
Diagnostic après la naissance
L'examen d'un enfant malade vise à identifier les malformations. Un nouveau-né atteint du syndrome d'Edwards est examiné par différents spécialistes :
- néonatologiste;
- cardiologue;
- neurologue;
- chirurgien;
- orthopédiste;
- urologue, etc...
Les études diagnostiques les plus importantes réalisées sur un enfant atteint du syndrome d'Edwards immédiatement après sa naissance :
- échocardiographie;
- Échographie des reins;
- Échographie des organes abdominaux.
Certaines caractéristiques du déroulement de la grossesse lors du port d'un enfant atteint du syndrome d'Edwards ne diffèrent pas. Il commence également à bouger en temps voulu, parfois même très actif, aucune toxicose supplémentaire n'est observée. Par conséquent, seuls les équipements de diagnostic modernes peuvent détecter la maladie pendant cette période. Si le bébé est encore né, toute sa courte vie sera consacrée au traitement constant des défauts concomitants.
Avis des médecins. Les filles atteintes du syndrome d'Edwards naissent plus, non pas parce qu'elles sont le plus souvent sensibles à une telle pathologie sur le 18e chromosome. La raison en est leur survie. On pense que la plupart des grossesses de garçons atteints de ce syndrome se terminent par un avortement spontané ou une mort fœtale intra-utérine.
Traitement
Chez ces enfants, les anomalies du développement et toutes sortes de malformations sont incompatibles avec la vie, de sorte que le traitement du syndrome d'Edwards est réduit à des soins symptomatiques. Elle vise à maintenir les fonctions physiologiques de base, à améliorer la qualité de vie et à la prolonger au maximum. La correction chirurgicale des pathologies congénitales, selon la plupart des médecins, est injustifiée et très risquée. Dans les premiers jours de la vie, le traitement est limité à des activités telles que :
- alimentation par sonde en raison de difficultés de déglutition et de succion ;
- IVL prolongé, car il y a des problèmes respiratoires.
Après la sortie (si possible), les parents doivent fournir à un bébé malade :
- soins organisés, presque professionnels : c'est bien s'il y a une personne (nounou) avec une formation médicale à côté de l'enfant ;
- nutrition complète;
- surveillance régulière par divers pédiatres.
Il faut tout de suite faire une réserve que s'occuper d'un enfant atteint du syndrome d'Edwards demande beaucoup de patience et de force.
Prévisions

Les parents dont l'enfant est porteur du syndrome d'Edwards doivent savoir quelles prévisions les attendent. Comme mentionné ci-dessus, ils sont très décevants :
- l'espérance de vie de ces enfants est très faible;
- 60% meurent avant 3 mois (le plus souvent les garçons entrent dans ce groupe) ;
- un autre 10% vivent jusqu'à un an;
- seulement 1 % des personnes nées vivent jusqu'à 10 ans ;
- la cause la plus fréquente de décès est l'insuffisance cardiaque ou l'arrêt respiratoire;
- tous les enfants atteints du syndrome d'Edwards restent profondément oligophréniques jusqu'à la fin de leurs jours ;
- le corps de ces enfants est très affaibli, sujet à toutes sortes de maladies, à cause desquelles ils souffrent souvent d'infections des voies urinaires, de conjonctivite, de moyen, de pneumonie;
- les compétences de communication verbale chez ces enfants sont très limitées;
- ils peuvent réagir aux paroles de leurs parents ;
- certains sourient même, reconnaissent et interagissent avec les soignants;
- capable d'acquérir des compétences telles que l'auto-alimentation et lever la tête.
Si pendant la grossesse l'enfant a été diagnostiqué avec le syndrome d'Edwards, cela oblige les parents à prendre une décision responsable (le moment du diagnostic de diverses pathologies fœtales peut être trouvé dans). Pourront-ils prendre en charge un enfant gravement malade souffrant de multiples pathologies ? Ou est-il judicieux d'interrompre une telle grossesse? Ici, seul le couple lui-même, par des efforts conjoints, peut trouver la seule bonne issue pour eux.
En 1971 Lors de la Conférence de Paris, une nomenclature spéciale pour l'enregistrement du caryotype humain a été approuvée.
Caryotype humain normal:
46,XX - femme; 46 ans, HU est un homme.
Caryotype en polyploïdie:
69,XXX ; 69, XXY - triploïdie ;
92,XXXX ; 92,XXXX - tétraploïdie.
Carytype avec monosomie :
45, XO est la seule monosomie possible chez les personnes vivantes (syndrome de Shereshevsky-Turner).
Caryotype dans les trisomies autosomiques:
47, XX, + 21 ou 47, XY, + 21 - trisomie sur le chromosome 21 (syndrome de Down);
47, XX, + 13 ou 47, XY + 13 - trisomie sur le chromosome 13 (syndrome de Patau);
47, XX + 18 ou 47, XY, + 18 - trisomie sur le chromosome 18 (syndrome d'Ewards).
Caryotype des chromosomes sexuels de la trisomie:
47,XXX - trisomie X chez une femme ;
47,XYU - trisomie Y chez un homme.
47, XXY - Syndrome de Klinefelter.
Tétrasomie et pentasomie sur les chromosomes sexuels:
48,ХХХХ - tétrasomie X ;
49,ХХХХХ - pentasomie X;
48,XXXX ; 49,ХХХХУ - variantes du syndrome de Klinefelter;
48,HUUU ; 49, HUUUU - variantes du syndrome de polysomie Y chez un homme.
Caryotype dans les aberrations chromosomiques:
46,XX,del 5p - - délétion du bras court du chromosome 5 (syndrome du cri du chat) chez une femme ;
46,XY,del 4p - - délétion du bras court du chromosome 4 (syndrome de Wolf-Hirschhorn) chez un homme ;
46, X, i (Xq) - isochromosome X le long du bras long d'une femme;
46, XY, r (18) - chromosome radial 18 chez un homme ;
45, XX, -D, -U, + t (Dq, Uq) - translocation Robertsonienne équilibrée formée par la connexion des bras longs d'un chromosome D et d'un chromosome Y chez une femme.
Caryotype mosaïque:
45,X /46,XX ou 45,X /46,XX - une partie des cellules a un caryotype normal (46,XX) et une partie avec une monosomie X (45,X). Nous parlons de la forme mosaïque du syndrome de Shereshevsky-Turner;
47,XX,+21/ 46,XX - forme mosaïque du syndrome de Down.
pathogenèse des maladies chromosomiques.
Dans les maladies chromosomiques, en règle générale, il existe un déséquilibre dans un grand nombre de gènes. Le génotype altéré se manifeste dans la période embryonnaire de développement. Les premières étapes du clivage du zygote sont contrôlées par des substances accumulées dans l'œuf. Ensuite, les propres gènes du zygote sont activés. Au total, environ 1000 gènes sont actifs dans la période embryonnaire, qui sont responsables des différentes étapes de l'ontogenèse. Ils sont dispersés sur tous les chromosomes. Avec les mutations génomiques et chromosomiques, l'équilibre est perturbé dans un grand nombre de gènes, y compris les gènes qui régulent le développement embryonnaire. Cela conduit inévitablement à une violation de l'histogenèse et de l'organogenèse. Des malformations se forment. Le plus souvent, les violations sont incompatibles avec la vie, ce qui entraîne la mort intra-utérine de l'embryon. Rarement, un enfant naît avec des malformations.
De 35 à 50% (maintenant ils écrivent jusqu'à 70%) des embryons humains meurent au stade blastocyste, c'est-à-dire avant implantation. Un grand pourcentage d'entre eux ont des réarrangements chromosomiques. Après l'implantation, la contribution totale des anomalies chromosomiques à la mort intra-utérine chez l'homme est de 45 % : plus une grossesse est interrompue tôt, plus elle est susceptible d'être due à un déséquilibre chromosomique.
Si un avortement survient au cours des 2 à 4 premières semaines, un déséquilibre chromosomique est observé dans 60 à 70% des avortements. Au premier trimestre - à 50%, au 2ème trimestre - à 30%, à 20-27 semaines - à 7% et, enfin, 6% des mortinaissances sont dues à une pathologie chromosomique.
Si les violations du développement embryonnaire sont compatibles avec la vie, un enfant naît avec des malformations.
1 % vivant les nouveau-nés sont atteints de certaines maladies chromosomiques.
Cliniquement, les maladies chromosomiques se manifestent par des syndromes de malformations congénitales multiples. Presque tous sont formés au moment de la naissance. Les exceptions sont les violations de la formation des caractéristiques sexuelles avec un déséquilibre des chromosomes sexuels. Certains de leurs symptômes apparaissent pendant l'adolescence. Les généticiens comparent les maladies chromosomiques aux cendres après un incendie. Le feu est ce qui se passe dans la période embryonnaire. Au moment de la naissance, le phénotype final est formé (tisons après le feu). Rien ne peut être réparé. Vous ne pouvez effectuer qu'une correction esthétique, opérer un patient présentant un défaut de développement (si le syndrome est compatible avec la vie).
Étant donné que les premiers stades du développement embryonnaire sont perturbés dans la bronchite chronique, de nombreux organes et systèmes d'organes sont touchés simultanément. Cela rend le tableau clinique de nombreuses maladies chromosomiques similaires. Plus le déséquilibre des chromosomes est important, plus l'image est non spécifique.
Pour toute maladie chromosomique, le polymorphisme est caractéristique, tk. le génotype individuel des individus affecte l'expression des gènes.
CARACTÉRISTIQUES CLINIQUES ET CYTOGÉNÉTIQUES DES MALADIES CHROMOSOMIQUES LES PLUS COURANTES