Erb miopatie juvenilă. Distrofia musculară progresivă Erb-Roth
Citeste si
În neurologia modernă, există un număr mare de boli, a căror natură apariției nu poate fi explicată rațional de către specialiști. Astfel de boli includ un grup de afecțiuni precum distrofia musculară. Există nouă varietăți ale acestei boli, dar mai întâi de toate...
Distrofia musculară este o cronică boala ereditara ca urmare a dezvoltării căreia sistemul muscular uman este deteriorat. Mușchii afectați încetează să funcționeze normal, devin mai subțiri în dimensiune și un strat de grăsime crește treptat la locul lor în corp.
Varietăți de distrofie musculară
În neurologia modernă, această boală este clasificată în nouă boli diferite. Clasificarea bolii este asociată cu:
- localizarea tulburărilor musculare;
- caracteristicile bolii;
- dezvoltare agresivă;
- vârstă.
Deci, se întâmplă distrofia musculară:
- Duchenne;
- miotonic (boala Steinert);
- Becker;
- Erba Roth;
- forma juvenilă de distrofie Erba-Roth;
- umăr - formă facială scapulară (Landuzi-Dejerine);
- miopatie alcoolică;
- forma distală;
- Miodistrofia Emery-Dreyfus.
Distrofia Duchenne
Cel mai formă cunoscută distrofie Duchenne progresivă (distrofie pseudohipertrofică, merozină - distrofie congenitală negativă). Acest tip de boală se manifestă în copilărie de la doi la cinci ani. În primul rând, mușchii extremităților inferioare suferă de această boală, care, în ciuda unui stil de viață sedentar la pacienții tineri, crește treptat în dimensiune. Această caracteristică este asociată cu o creștere a țesutului adipos în loc de mușchi.

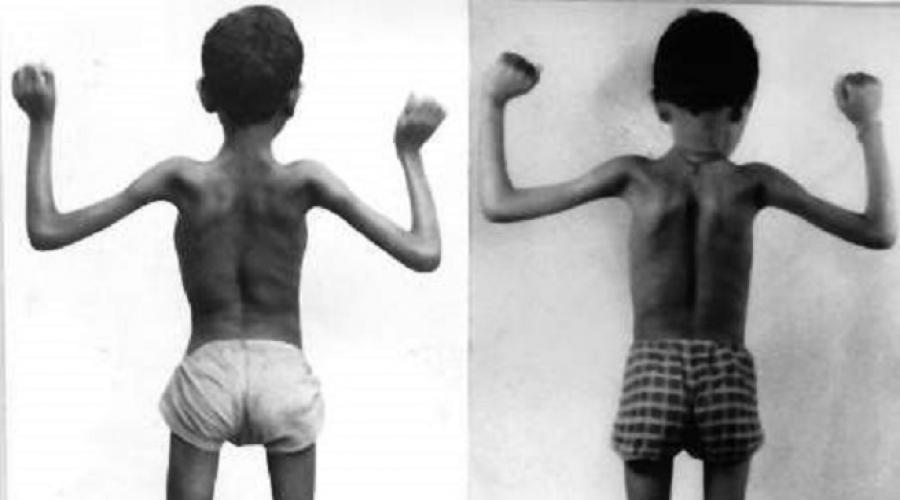
Copil sănătos în stânga, copil bolnav în dreapta
Treptat, pe măsură ce boala progresează, se deplasează în partea superioară și afectează mușchii membrelor superioare. De regulă, până la vârsta de doisprezece ani, un pacient mic încetează complet să se miște. Letalitatea acestei boli este foarte mare, aproximativ 85-90% dintre pacienți nu trăiesc până la 20 de ani.
Bărbații sunt expuși riscului, deoarece această boală practic nu afectează fetele.
boala Steinert
Nu în zadar distrofia congenitală a lui Steiner se numește miotonică, deoarece, ca urmare a progresiei bolii, mușchii se relaxează prea încet după contracția lor (acest fenomen se numește miotonie).
Această boală, spre deosebire de cea anterioară, este frecventă la adulții cu vârsta cuprinsă între 20 și 40 de ani. Există și cazuri de progresie a bolii la copii, de obicei în copilărie, cu toate acestea, aceasta este mai degrabă excepția decât regula.

Semne faciale de miotonie (gura deschisă și pleoapele)
Boala nu are o dependență de gen și afectează în egală măsură atât bărbații, cât și femeile. Se remarcă faptul că, cu o boală, se manifestă slăbiciune a mușchilor faciali ai feței, precum și a membrelor. Progresia, spre deosebire de boala Duchenne, este lentă.
O trăsătură distinctivă a bolii este posibilitatea de deteriorare nu numai a mușchilor membrelor, ci și a mușchilor interni (mușchiul inimii), care, la rândul său, prezintă un mare pericol pentru viața umană.
boala Becker
Această formă a bolii progresează, de asemenea, pentru o lungă perioadă de timp, iar pacientul perioadă lungă de timp se simte bine. Exacerbarea bolii poate apărea pe fondul leziunilor sau diverse boli sistem nervos care, prin cursul lor, vor accelera dezvoltarea bolii.
La risc sunt persoanele de statură mică.
Boala Erb-Roth și forma sa tinerească
Această boală autozomal recesivă se dezvoltă la pacienți după vârsta de 20 de ani, iar forma tinerească la copii și adolescenți cu vârsta cuprinsă între 11-13 ani. Progresia bolii are loc într-o variantă ascendentă, adică mușchii extremităților inferioare sunt mai întâi afectați, iar boala urcă treptat către extremitățile superioare. 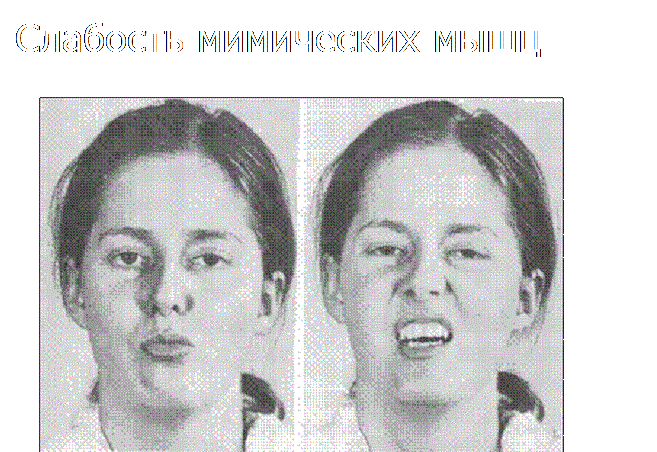
O trăsătură distinctivă a bolii este prezența omoplaților proeminenti, care, pe măsură ce progresează, devin mai pronunțate și mai evidente.
În timpul mersului, se notează transbordarea pacientului, proeminența abdomenului și retragerea toracelui.
boala Landouzy-Dejerine
Forma omoplatului-facială a acestei boli este cea mai nediscriminatorie, deoarece afectează persoanele cu vârsta cuprinsă între cinci și 55 de ani. Această boală se caracterizează prin dezvoltare îndelungată, pacientul poate rămâne capabil să lucreze până la 25 de ani de viață cu această boală.
Simptomele distinctive sunt afectarea mușchilor faciali, în urma cărora pacientul poate avea probleme cu claritatea pronunției, din cauza închiderii incomplete a buzelor. În plus, se observă închiderea incompletă a pleoapelor.
Pe măsură ce pacientul se dezvoltă, mușchii faciali, mușchii umerilor, membrelor și trunchiului se atrofiază.
Această formă nu depinde de o mutație genetică.
Miopatie alcoolică
De asemenea, acest tip de boală nu este asociat cu mutații genetice și există un singur motiv pentru apariția sa - abuzul de alcool. Pacienții pot prezenta sindroame dureroase la nivelul extremităților asociate în mod specific cu leziuni musculare.
Există miopatie alcoolică acută și cronică.
Forma distală
Forma distală a distrofiei musculare este o variantă benignă a distrofiei progresive. De regulă, această boală este dificil de diferențiat de amiatrofia neuronală a lui Marie-Charcot. Pentru a separa aceste două boli, este necesară o electroencefalogramă a capului, care oferă o înțelegere a bolii care trebuie tratată.
Principalele simptome ale bolii includ atrofia mușchilor membrelor cu emaciarea lor ulterioară. Posibilă pareză a picioarelor, mâinilor etc.
Miodistrofia Emery-Dreyfus
Acest tip de boală nu a fost inițial identificat ca o boală separată, deoarece în simptomele sale era similar cu distrofia Duchenne. Cu toate acestea, mai târziu, în urma unui studiu pe termen lung, s-a constatat că boala Emery-Dreyfus are simptome individuale.
Boala este clasificată ca fiind rară. La risc sunt persoanele sub 30 de ani, de obicei o vârstă fragedă. Există dovezi ale manifestării simptomelor după 30 de ani, dar acestea sunt rare.
Principala diferență între acest tip de boală este problemele cu mușchii inimii, care în cele din urmă pot provoca moartea. Cardiomiopatia în această boală nu este singura diferență. Pe lângă problemele cardiace, pacienții au semne standard pentru distrofia Duchenne, dar într-un mod de dezvoltare mai benign.
Cauze
Componenta negativă a bolilor sistemului nervos este că sunt greu de studiat. Din acest motiv, nu se cunosc pe deplin factorii care provoacă dezvoltarea unei forme sau alteia de distrofie musculară.
Motivul principal pentru formarea majorității subspeciilor acestei boli este o mutație genetică, în special gena care este responsabilă de sinteza proteinelor.
De exemplu, boala Duchenne este direct legată de mutația cromozomului X sexual. Principalul purtător al unei gene rele sunt fetele care, în ciuda prezenței acesteia în propriul ADN, nu suferă de această boală.
În ceea ce privește forma miotonică, vinovată de apariția acesteia este o genă situată pe cromozomul 19.
Principalele simptome
Prezența unui număr mare de subspecii diferite în această boală indică diferențe de simptome, cu toate acestea, boala are simptome comune, care includ:

Diagnosticul distrofiei musculare
Măsurile de diagnosticare pentru o astfel de boală sunt extinse, deoarece există un numar mare de boli de care este necesară diferențierea bolii.
În stadiul inițial, medicul va studia cu siguranță istoricul pacientului, va clarifica simptomele, rutina zilnică etc. Aceste date sunt necesare pentru a elabora un plan pentru măsurile de diagnostic ulterioare, care pot include:

Este de remarcat faptul că, cu cât boala s-a manifestat mai târziu, cu atât mai bine pentru pacient, deoarece simptome precoceîn majoritatea cazurilor se termină cu moartea.
Tratament
Tratamentul distrofiei musculare este un proces complex și îndelungat, totuși, în prezent, nu a fost creat un medicament care să vindece complet pacientul. Toate activitățile sunt menite să ușureze viața pacientului și să restabilească unele dintre abilitățile pierdute.
Pentru a încetini dezvoltarea bolii, sunt prescrise următoarele medicamente:
- corticosteroizi;
- vitaminele B1;
- adenozin trifosfat (ATP).
În plus, pentru a încetini procesul de dezvoltare, se folosesc celule stem fetale, care încetinesc procesul de distrofie.
În plus, ca măsuri preventive numi:
- masaj;
- fizioterapie;
- exerciții de respirație.
Pe lângă opțiunile de tratament standard, este important să fii ghidat în mod constant de trei componente principale în procesul vieții:
- Activitate fizică adecvată.
- Suport psihologic în timp util.
- Tine dieta.
Activitate fizica
Lipsa dorinței unei persoane de a lupta împotriva bolii are impact negativ pe corp. Judecă singur, pasivitatea, lipsa de dorință de a se mișca îl deprimă pe cei deja afectați sistem muscular. Muschii trebuie să primească o sarcină, deoarece fără încărcare, procesele distrofice încep să apară mai repede, progresând astfel într-un ritm mai rapid.
Activitate fizică moderată, utilizarea dispozitivelor de susținere va fi un ajutor excelent în lupta împotriva bolii.
În prezența durerii în mușchi, înotul, yoga, exercițiile de întindere sunt perfecte.
Suport psihologic
Suportul psihologic al mediului este important pentru o persoană bolnavă. Și dacă boala este la fel de gravă ca aceasta, cu atât mai mult. Pentru unii, sprijinul obișnuit din partea prietenilor și familiei va fi suficient, în timp ce alții pot avea nevoie de ajutor psihologic calificat.
Este important să-i spunem clar unei astfel de persoane că nu a rămas singur cu problema lui. Trebuie să înțeleagă că are la cine să apeleze, sunt oameni care îl empatizează și îl susțin.
Cura de slabire
În ceea ce privește dietă și dietă, există o credință comună că respectarea unei diete antiinflamatorii poate încetini progresia bolii. Această dietă reduce inflamația, nivelul de glucoză, elimină toxinele din organism și îl hrănește cu substanțe benefice.
Esența unei astfel de diete este următoarea:
- Refuzul produselor care conțin grăsimi „rele” și înlocuirea lor cu altele „bune”, introducerea în alimentație a grăsimilor nesaturate, care se găsesc în măsline, semințe de in, ulei de susan, avocado.
- Utilizarea cărnii și a peștelui în alimente, în producția cărora nu s-au folosit antibiotice sau hormoni.
- Eliminarea completă a zahărului rafinat și a glutenului din dietă.
- Mananca urmatoarele alimente - varza chinezeasca, broccoli, telina, ananas, somon, sfecla, cucumaria, ghimbir, turmeric si alte alimente care au proprietati antiinflamatorii.
- Produsele lactate sunt permise numai pe bază de lapte de oaie și capră.
- Este permisă utilizarea ceaiurilor din plante, limonadă, kvas, băuturi din fructe și sucuri naturale.
Prevenirea
Deoarece distrofia musculară este destul de dificil de prezis și detectat într-un stadiu incipient, măsurile preventive se reduc la două recomandări simple:
Examinarea obligatorie a unei femei în etapa de planificare a sarcinii pentru prezența mutațiilor în gene
Dacă, dintr-un motiv oarecare, nu a fost efectuat un examen genetic înainte de sarcină, un test este efectuat deja în timpul sarcinii pentru a determina mutațiile cromozomului X la făt.
Prognostic și complicații
În funcție de tipul de boală, prognosticul poate fi diferit și totuși există mai multe posibile complicații care apar cu această boală.
- tulburări cardiace
- rahiocampsis
- scăderea abilităților intelectuale ale pacientului
- scăderea activității motorii
- tulburări ale sistemului respirator
- rezultat letal
Deci, distrofia musculară este o boală destul de periculoasă și incurabilă, așa că viitorii părinți trebuie să fie profund responsabili pentru planificarea sarcinii. Ai grijă de tine și de viitorii tăi copii!
Insistați asupra unui diagnostic precis, studiați informațiile, iar articolul Claudiei vă va ajuta în acest sens.
Diagnosticul meu este miopatie Erb-Roth! Este corect?
De ce am nevoie de un diagnostic precis? Este inca incurabil...
De ce am nevoie de diagnostice suplimentare? Diagnosticul meu este miopatie Erb-Roth!
Am avut deja o grămadă de teste și totul a fost negativ acolo...
Sunteți familiarizat cu astfel de gânduri?
După cum am scris de mai multe ori, miopatia Erb-Roth (sau LMMD) este un diagnostic descriptiv. Sub acest nume se ascund zeci de boli. caracteristici diferite, prognostic și potențial de tratament.
Și printre ele există un diagnostic care trebuie abordat Atentie speciala. Aceasta este până acum singura distrofie musculară a centurii membrelor pentru care există un remediu. Acest LGMD este o boală Pompe cu debut tardiv.
Este inutil să ne complați într-o descriere a manifestărilor tipice ale bolii Pompe: există atât de multe fețe în această boală. Cineva are activitatea CPK (creatina kinazei) la limita superioară a normei (aproximativ 300 de unități / l), iar pentru cineva - sub 3000 de unități / l. La un pacient, boala începe cu dificultăți de respirație și apnee în somn, în timp ce la altul, mușchii scheletici suferă mai mult, dar totul este în regulă cu respirația. Boala Pompe cu debut tardiv se poate manifesta la 5 ani și la 55 de ani.
Dacă ați fost diagnosticat cu distrofie musculară a membrelor sau cu miopatie Erba-Roth, este posibil să aveți boala Pompe cu debut tardiv.
Fă-ți testul gratuit pentru boala Pompe. Pentru a face acest lucru, trebuie să găsiți un medic de orice specialitate care va fi de acord să vă ajute. Acest doctor are nevoie de un telefon linia fierbinte suport pentru diagnosticarea bolii Pompe - 88001002494 (despre programul de diagnostic de pe site-ul Centrului Științific de Stat din Moscova).
Informații suplimentare: .Analiza se face pe pete de sânge uscat. Specialistul liniei fierbinți vă va spune cum să faceți acest lucru și de unde să obțineți materialele necesare.
În primul rând, va fi efectuat un test enzimatic de screening pentru a vedea dacă ați fost exclus pentru boala Pompe. Dacă rezultatul arată că nu este exclus, atunci laboratorul va efectua o analiză ADN și numai rezultatul acestuia va pune capăt întrebării prezenței bolii Pompe.
La Moscova, acest lucru se poate face, de exemplu, la Centrul de Cercetare Genetică Medicală de pe stradă. Moskvorechye, 1. Dacă nu locuiți la Moscova și doriți să excludeți boala Pompe, atunci scrieți Inna Stanovoy- cea mai activă pacientă rusă cu boala Pompe, vă va spune cum să acționați.
Medicamentul pentru tratamentul bolii Pompe - Myozyme - este înregistrat în Rusia, aproape toți pacienții ruși cu boala Pompe îl primesc gratuit. Obținerea acestuia nu este ușor, dar merită, deoarece Myozyme este o terapie patogenetică, adică una care acționează asupra cauzei bolii Pompe, încetinind sau oprind foarte mult dezvoltarea acesteia, și nu doar atenuând simptomele.
Nu pierde timpul! Ce se întâmplă dacă ai boala Pompe?
Miodistrofie- Atrofie musculară progresivă. Se observă destul de des. Persoanele de ambele sexe se îmbolnăvesc. Boala este ereditară. Există mai multe forme de miodistrofie, care sunt moștenite conform tipuri diferiteși de aceea sunt considerate boli independente. Dintre acestea, cea mai frecventă este miodistrofia juvenilă Erba-Roth, miodistrofia omoplată-facială a lui Landouzy-Dejerine și miodistrofia pseudohipertrofică a lui Duchenne.
Distrofia musculară juvenilă a lui Erb - Rota
Distrofia musculară juvenilă a lui Erb - Roth se moștenește într-o manieră autosomal recesivă (copiii se îmbolnăvesc parinti sanatosi). Primele semne ale bolii apar în principal la vârsta de 14-16 ani, foarte rar - la vârsta de 5-10 ani. Simptomele inițiale sunt slăbiciune musculară, oboseală musculară patologică cu activitate fizica; mersul devine „rață”. Atrofiile sunt mai întâi localizate în grupele musculare proximale ale centurii pelvine și ale extremităților inferioare. Uneori, procesul miodistrofic afectează simultan mușchii pelvini și ai centurii scapulare. În etapele ulterioare, mușchii spatelui și abdomenului sunt implicați în proces. Ca urmare a atrofiei, se formează lordoza, scapula pterigoidiană, „talie de viespe”. Când se ridică, pacienții folosesc tehnici auxiliare, își sprijină mâinile pe șolduri. Manifestarea pseudohipertrofiei musculare, contractura articulara, retractia tendonului este nesemnificativa. Reflexele tendinoase de la extremitățile superioare și inferioare sunt reduse. Cursul bolii este lung și progresiv. La vârsta de 35-40 de ani, pacienții își pierd capacitatea de a se mișca independent, dizabilitățile se instalează devreme.Miodistrofie umar-scapulo-facial Landuzi - Dejerine
Miodistrofia umar-scapulo-faciala Landuzi - Dejerine se mosteneste in mod autosomal dominant.Manifestări clinice ale miodistrofiei umăr-scapulare-faciale Landuzi - Dezhsrina
Primele semne ale bolii apar mai ales la vârsta de 10-20 de ani. Slăbiciunea și atrofia musculară sunt localizate în principal pe față, omoplați și umeri. Din cauza atrofiei, fața devine hipomimică. Pentru pacienți, sunt tipice o frunte netedă, lagoftalmia, un zâmbet „transvers”, buzele groase, uneori inversate (buzele unui tapir). Atrofia mușchilor umărului, pectoralului mare, sertului anterior, mușchilor trapezi provoacă apariția simptomelor de suprafață laxă, scapulelor pterigoide, o creștere a spațiului interscapular, aplatizarea toracelui,. Uneori, atrofia se extinde la mușchii extremităților inferioare. Pseudohipertrofie exprimată în mușchii gambei și deltoizi. ÎN primele etape boală, tonusul este redus în grupele musculare proximale. Reflexele tendinoase sunt reduse (în principal cu mușchii bicepși și tricepși ai umărului). Cursul bolii este lent progresiv. Pacienții rămân funcționali pentru o lungă perioadă de timp.Miodistrofie pseudohipertrofică Duchenne
Miodistrofia Duchenne pseudohipertrofică este un malign al tuturor bolilor sistemului neuromuscular. Această boală se observă numai la băieți, deci este moștenită într-un tip recesiv, legat de X. Boala se transmite pe linie maternă, începe în primii ani de viață. Atrofia musculară este inițial localizată în mușchii centurii pelvine și ai coapselor, drept urmare dificultățile apar devreme la urcarea scărilor; mersul devine „rață”. Copiii cad adesea și se ridică cu dificultăți. Schimbările statice. La vârsta de 10-12 ani, pacienții își pierd capacitatea de a se mișca independent, sunt imobilizați la pat. Cu această formă de miodistrofie, inteligența este afectată. Există modificări în mușchii inimii. Se dezvoltă pseudohipertrofia mușchilor gambei. Baza oricărei miodistrofii progresive este degenerarea treptată a fibrelor musculare. mușchi scheleticși înlocuirea acestora cu țesut conjunctiv și adipos. Ca urmare, se dezvoltă falsă hipertrofie musculară, mai des gastrocnemiul (cu miodistrofie Duchenne și Erb) și retracția (contracția) tendoanelor lui Ahile. Degenerarea musculară apare din cauza metabolismului alterat patologic în ei. Metabolismul proteinelor și carbohidraților este grav perturbat.Tratamentul miodistrofiei Duchenne pseudohipertrofice
Recomand o dieta bogata in proteine (carne, peste, branza), vitamine. Nu este necesar să se limiteze mișcările membrelor superioare, dimpotrivă, fizioterapie si masaj. Retabolil se administrează în cure (1 ml pe săptămână, 4 injecții în total). ATP este prezentat la 1 ml intramuscular zilnic (pentru un curs de tratament 15-20 injecții), cerebrolizină (1 ml intramuscular, 20-300 injecții), precum și anaprilină - 20-40 mg de 2 ori pe zi (timp de 4 săptămâni cu întreruperea treptată a medicamentului). De asemenea, se recomandă să luați acid glutamic, riboxină, metionină. Se prescriu tocoferol, retinol, acid ascorbic, vitaminele B. Sunt prezentate mijloace care îmbunătățesc microcirculația: acid nicotinic, nicotinat de xantinol, sermyun, actovegin, pentoxifilină, parmidină. Pentru a îmbunătăți conducerea neuromusculară, se folosesc medicamente anticolinesterazice: prozerină, galantamina, oxazil, bromură de piridostigmină, sulfat de stefaglabrină. Numiți simultan exerciții de fizioterapie, masaj, fizioterapie, acupunctura. Importantă este prevenirea deformărilor osteoarticulare și a contracturilor membrelor. Se folosesc procedee termice: ozocerită, aplicații cu nămol, radon, conifere, băi sulfurate, hidrogen sulfurat, baroterapie cu oxigen. Tratament ortopedic indicat ptDistrofia musculară progresivă Erb-Roth este una dintre cele forme ereditare afectarea țesutului muscular.
Debutul bolii este de obicei la vârsta de 10-20 de ani (debutul este posibil până la 40 de ani); imobilizarea completă durează aproximativ 10-15 ani. Transmiterea trăsăturii se face după tipul recesiv, legat de podea.
Informații pentru medici. Distrofia musculară Erb-Roth este criptată conform ICD 10 sub codul G71.0. În același timp, stadiul bolii trebuie indicat în diagnostic (1 - tulburări moderate de mișcare, 2 - apar dificultăți la mers, la efectuarea fizică ușoară munca, 3 - paralizii, contracturi etc.). După aceea, se indică severitatea manifestărilor concomitente (scăderea inteligenței, complicații de la inimă), rata de progresie (rapidă, medie sau lentă). În caz de pierdere a capacității de mișcare, acest fapt trebuie indicat.
Cauze
Nu există un punct de vedere unic asupra apariției atrofiei musculare. Teoria ereditară domină. Mecanismele biochimice exacte ale patogenezei nu au fost pe deplin elucidate.
Simptome
Simptomele bolii nu sunt specifice la început și includ slăbiciune generală, slăbiciune a mușchilor spatelui. Treptat, boala progresează. Pacientul încetează să-și țină spatele într-o poziție normală, se dezvoltă hiperlordoză - supraextensia posterior a spatelui inferior.
Mersul se schimbă destul de devreme. Devine ca o „răță” - picioarele cântărite din cauza slăbiciunii mușchilor coapsei și ai centurii pelvine. Hipotrofia mușchilor centurii scapulare superioare se dezvoltă rapid. De asemenea, hipotrofia generală se dezvoltă treptat, apoi atrofia musculară. Uneori are pseudohipertrofie a picioarelor - înlocuire masa muscularațesut adipos și conjunctiv.
În timp, pacientul încetează să efectueze multe mișcări active. Ridicarea este mult mai dificilă, pacienții trebuie să se ridice în patru picioare, să se ajute cu mâinile când se ridică. Fața pacientului devine amimic, pleoapele nu se închid complet, iar buzele, dimpotrivă, se întorc în față și deseori se îngroașă (buze tapir). Expresiile faciale ale unui astfel de pacient sunt uneori numite și chipul unui miopat.
Diagnosticare
Diagnosticul bolii este de obicei stabilit destul de precis. Când se face un diagnostic de distrofie Erba-Roth, se acordă atenție vârstei la debutul bolii, eredității și ritmului de progresie a procesului. Un examen neurologic relevă o scădere a reflexelor până la pierdere, o scădere a tonusului muscular, prezența contracturilor articulare (datorită procesului inegal de atrofie musculară).
Contrar concepțiilor greșite, contracțiile musculare fasciculare nu apar. La înregistrarea biocurenților musculari, amplitudinea scade, dar nu și frecvența descărcărilor. Conform ENMG, scurtarea duratei potențialelor de acțiune, se determină înregistrarea polifazică.
Biochimic, este adesea detectată o modificare a activității creatininei kinazei, AST și a altor enzime. Uneori este detectată o modificare a compoziției electroliților din sânge. 
Diagnosticul este considerat de încredere atunci când se efectuează un examen histologic al mușchilor. Forma și dimensiunea fibrelor musculare se modifică, se modifică percepția culorii lor de către coloranții histologici, mușchii renasc, volumul nucleilor musculari crește. Între fibrele musculare se determină grăsimea, țesut conjunctiv. În același timp, nu există o distribuție a fasciculului de fibre, ceea ce este caracteristic miopatiilor neurogenice.
Tratament
Terapia patogenetică nu a fost dezvoltată. Tratamentul este simptomatic și urmărește reducerea ratei de progresie. Utilizați activ vitaminele B, vitamina E, ATP, extract de aloe intramuscular, ATP. Cu ceva timp în urmă, a fost folosit hormonul anabolic retabolil, dar s-a observat adesea o creștere a degradarii țesutului muscular. De asemenea, se utilizează medicamente precum acidul tioctic, riboxină, actovegin.
Un rol important este acordat metodelor de expunere non-medicamentale. Masajul pentru pacienții cu distrofie Erba-Roth trebuie efectuat într-un ritm ușor, având ca scop combaterea spasmului muscular, întărirea mușchilor. De asemenea rol important aparține LFC. Terapia cu exerciții fizice pentru boală ar trebui să fie moderată, dar regulată, ideal zilnic. Toate grupele musculare sunt antrenate.
Constanța măsurilor preventive permite pentru o lungă perioadă de timp menținerea capacității pacienților de autoservire. În practica mea, îmi amintesc un pacient care, la debutul bolii la vârsta de 25 până la 60 de ani, și-a păstrat capacitatea de a se deplasa independent, în autoservire, deși cu o limitare.
Prognoza
Prognosticul pentru toate distrofiile musculare este de obicei prost. Boala progresează treptat, acoperind toate grupele musculare. Mai devreme sau mai târziu, are loc imobilizarea pacientului. Deși boala în sine practic nu duce la moartea pacientului. Moartea se datorează escarelor, infecțiilor pulmonare, tractului urinarși așa mai departe.
Se moștenește în mod autosomal recesiv (copiii părinților sănătoși se îmbolnăvesc).Dezvoltare: Boala debutează la vârsta de 14-18 ani. Atrofia (distrofia) mușchilor centurii pelvine se dezvoltă treptat, ducând la o schimbare a mersului, care seamănă cu o „răță”. Apoi procesul se extinde la mușchii centurii scapulare și la mușchii spatelui.
Simptome: Pacienții au dificultăți să se ridice de la podea, ajutându-se cu mâinile; urcă treptele scărilor, sprijinindu-și mâinile pe balustradă. Slăbiciunea și atrofia mușchilor centurii scapulare fac dificilă ridicarea brațelor în sus. Atrofia mușchilor care fixează omoplații duce la dezvoltarea omoplaților pterigoidieni. Mișcările mâinilor și mușchilor feței nu sunt perturbate. Este posibilă creșterea mușchilor gambei picioarelor, care devin denși la atingere, dar puterea lor este redusă. Aceasta este pseudohipertrofia mușchilor gambei, care se dezvoltă ca urmare a creșterii țesutului conjunctiv și adipos în ei. Intelectul pacienților nu suferă.
Cursul bolii este lung. Până la vârsta de 35-40 de ani, pacienții își pierd capacitatea de a se mișca independent.
MIOPATIE DUCHENNE PSEUDOHIPERTROFĂ
Cea mai malignă dintre toate bolile sistemului neuromuscular.Această boală se observă numai la băieți, adică este moștenită legată de cromozomul sexual. Boala se transmite prin linie maternă.
Dezvoltare: Boala începe în copilărie, la vârsta de 3-5 ani, procesul atrofic captează mușchii centurii pelvine și coapselor, drept urmare dificultățile apar devreme la urcarea în deal, la urcarea scărilor, mersul devine o rață. . Copilul cade adesea, se ridică cu dificultate. Schimbările statice. Până la vârsta de 10-12 ani, pacienții cu miopatie Duchenne își pierd capacitatea de a se mișca independent și devin imobilizați la pat.
Cu această formă de miopatie, intelectul poate suferi. Găsiți modificări ale mușchiului inimii. Se dezvoltă pseudohipertrofia mușchilor gambei. La baza tuturor distrofiilor musculare progresive se află moartea treptată a fibrelor musculare ale mușchilor scheletici și înlocuirea lor cu țesut conjunctiv și adipos. Ca urmare, se dezvoltă pseudohipertrofia mușchilor, mai des mușchii gambei (cu miopatii Duchenne și Erb) și retracția (scurtarea) tendoanelor lui Ahile. Moartea musculară apare ca urmare a unui metabolism alterat patologic despre ei. Metabolismul proteinelor și carbohidraților este grav perturbat.
Tratament: O dietă bogată în proteine (pește, carne, brânză de vaci) și vitamine. Nu trebuie să vă limitați mișcările, dimpotrivă, sunt afișate exerciții terapeutice și masaj. Injecțiile cu Retabolil se efectuează în cure (1 injecție pe săptămână, 4 injecții în total), ATP (15-20 injecții pe curs), cerebrolysip (1 ml intramuscular, 20-30 injecții), precum și anaprilină (obzidan) 20- 40 mg de 2 ori pe zi (în decurs de 4 săptămâni, urmată de întreruperea tratamentului cu o reducere a dozei zilnice de 10 mg). De asemenea, se recomandă să luați acid glutamic (0,5 g de 3 ori pe zi), exazil (1 comprimat de 2 ori pe zi). Este posibil să se efectueze transfuzii fracționate de sânge dintr-un singur grup de 250 ml. Cursurile de tratament durează 4-6 săptămâni. cu pauză de 3-5-12 luni. in functie de evolutia bolii.
DISTROFIE MUSCULARĂ PROGRESIVĂ LANDUZY-DEJERINE
Descris pentru prima dată de Landouzy și Dejerine în 1885. Boala este a doua cea mai frecventă dintre distrofiile musculare progresive și apare cu o frecvență de 1:20.000 de persoane.feluri:
1) tineret clasic tip 1A (OMIM: 158900);
2) copilăria timpurie;
3) scapulohumeral fără implicarea mușchilor faciali (OMIM: 600416).
Simptome și dezvoltare:
1) Vârsta de debut a variantei clasice a bolii variază de la 10 la 20 de ani. Primele semne de slăbiciune musculară apar în mușchii mimici, dând feței pacientului un aspect specific ca de mască („fața sfinxului”). Buzele pacienților se îngroașă și ies în afară ("buze tapir"), există dificultăți în încrețirea frunții, îndoirea buzelor într-un tub, fluierat. Adesea, atunci când mușchiul circular al ochiului este implicat în proces, pacienții nu își pot închide strâns pleoapele, ceea ce este cel mai vizibil în timpul somnului.
Mușchii bulbari, extraculari și respiratori nu sunt de obicei afectați.
La unii pacienți, se observă aplazia congenitală a unei părți sau a întregului mușchi (de exemplu, m.pectoralis), a cărei etiologie nu este clară. De asemenea, se presupune că primele semne de slăbiciune musculară pot fi observate la nivelul mușchilor abdominali, ceea ce este rar observat atât de pacienți, cât și de medici.
Odată cu înfrângerea mușchilor faciali în stadiile incipiente ale bolii, se observă slăbiciune a mușchilor brâului scapular. Cei mai afectați sunt mușchii trapez, romboidal, pectoral și dorsal mare. Acest lucru duce la o limitare a amplitudinii de mișcare a articulației umărului (apar dificultăți la ridicarea brațelor deasupra nivel orizontal), și apariția așa-numitelor „vane pterigoide”. Implicarea în procesul mușchilor centurii pelvine și a grupelor musculare proximale ale picioarelor se observă doar la un mic procent de pacienți și numai în stadiile ulterioare ale bolii (de obicei după 15-20 de ani de la debutul primelor semne). a bolii). Pe membrele inferioare cei mai afectati sunt muschii iliaci, gluteali si adductori ai coapselor. O trăsătură caracteristică a bolii este asimetria leziunii, care poate fi observată chiar și în cadrul aceluiași grup muscular. Pseudo-hipertrofia mușchilor nu este caracteristică, dar poate apărea în stadiul târziu al bolii.
De asemenea, a fost descrisă prezența pacienților cu o combinație de simptome de LLP PMD cu hipoacuzie neurosenzorială și anomalie a capilarelor retiniene.
Boala progresează lent, fără a duce la dizabilitate severă și speranța de viață și fertilitatea reduse a pacienților.
2) Forma copilăriei timpurii a bolii apare la vârsta de 3 până la 6 ani și progresează rapid, ducând la imobilitate până la vârsta de 15-20 de ani.
Manifestările clinice sunt similare cu cele ale varianta clasica boli. Această formă se caracterizează prin apariția contracturilor, pseudohipertrofia mușchilor, precum și a scoliozei în regiunea toraco-lombară.
Sunt descriși pacienții cu debut precoce al bolii în combinație cu oligofrenie, epilepsie, surditate neurosenzorială și patologia vasculară retiniană.
Cel mai probabil, această variantă clinică nu este o formă nosologică independentă, deoarece a fost descrisă o combinație de variantă juvenilă și copilărie timpurie în aceeași familie. Se presupune că formele clasice și cele din copilărie timpurie ale bolii sunt variante alelice ale distrofiei musculare faciale-umăr-scapulare.
3) A treia opțiune este o formă rară de distrofie musculară progresivă scapulo-brahială care apare fără afectarea mușchilor faciali. Boala apare la vârsta de 12-40 de ani, se caracterizează printr-o progresie moderată și se moștenește în mod autosomal dominant.