Trisomia completă a cromozomului 7. Vezi ce este „cromozomul al 7-lea uman” în alte dicționare
Citeste si
Un grup special de boli asociate cu modificări structurale ale materialului genetic constă din bolile cromozomiale, clasificate condiționat ca fiind ereditare. Cert este că, în marea majoritate a cazurilor, bolile cromozomiale nu sunt transmise descendenților, deoarece purtătorii lor sunt cel mai adesea infertili.
Bolile cromozomiale sunt cauzate de mutații genomice sau cromozomiale care au apărut în gametul unuia dintre părinți sau într-un zigot format din gameți cu un set normal de cromozomi. În primul caz, toate celulele copilului nenăscut vor conține un set de cromozomi anormal (o formă completă a unei boli cromozomiale), în al doilea, se dezvoltă un organism mozaic, doar o parte a celulelor având un set anormal de cromozomi. (o formă mozaică a bolii). Severitatea semnelor patologice în forma mozaică a bolii este mai slabă decât în forma completă.
Baza fenotipică a bolilor cromozomiale este formată din încălcări ale embriogenezei timpurii, în urma cărora boala este întotdeauna caracterizată de malformații multiple.
Frecvența tulburărilor cromozomiale este destul de mare: din 1000 de bebeluși născuți vii, 3-4 au boli cromozomiale, la copiii născuți morți reprezintă 6%; aproximativ 40% dintre avorturile spontane sunt cauzate de un dezechilibru al cromozomilor (N.P. Bochkov, 1984). Numărul de variante ale bolilor cromozomiale nu este atât de mare pe cât s-ar putea aștepta teoretic. Un dezechilibru care afectează toate perechile de cromozomi provoacă tulburări atât de semnificative în organism încât acestea, de regulă, se dovedesc a fi incompatibile cu viața deja în stadiile incipiente sau ulterioare ale embriogenezei. Deci, monoploidia nu a fost găsită nici la nou-născuți, nici la avorturi. Sunt descrise cazuri rare de triploidie și tetraploidie la avorturi și la născuți vii, care totuși au murit în primele zile de viață. Modificările numărului sau structurii cromozomilor individuali sunt mai frecvente. Lipsa materialului genetic provoacă defecte mai semnificative decât un exces. Monosomia completă, de exemplu, pe autozomi practic nu este găsită. Aparent, un astfel de dezechilibru provoacă un rezultat letal deja în gametogeneză sau în stadiul de zigot și blastula timpurie.
Baza dezvoltării bolilor cromozomiale asociate cu o modificare a numărului de cromozomi se formează în gametogeneză, în timpul primei sau celei de-a doua diviziuni meiotice sau în timpul zdrobirii unui ovul fertilizat, cel mai adesea ca urmare a nedisjuncției cromozomiale. Mai mult decât atât, unul dintre gameți în loc de un singur set de cromozomi conține extrem de rar - un set diploid de toți cromozomii, sau 2 cromozomi din oricare dintre perechile de cromozomi, al doilea gamet nu conține niciun astfel de cromozomi. Când un ovul anormal este fertilizat de un spermatozoid cu un set normal de cromozomi sau un ovul normal de un spermatozoid anormal, mai rar atunci când sunt combinați doi gameți care conțin un număr modificat de cromozomi, sunt create condițiile prealabile pentru dezvoltarea unei boli cromozomiale.
Probabilitatea unor astfel de tulburări și, în consecință, nașterea copiilor cu boli cromozomiale, crește odată cu vârsta părinților, în special a mamei. Astfel, frecvența nedisjuncției celei de-a 21-a perechi de cromozomi din prima diviziune meiotică este de 80% din toate cazurile acesteia, din care 66,2% la mamă și 13,8% la tată; riscul total de a avea un copil cu trisomie pe cromozomul 13, 18, 21 pentru o femeie cu vârsta de 45 de ani și peste este de 60 de ori mai mare decât riscul pentru o femeie de 19-24 de ani (N.P. Bochkov și colab. 1984).
Sindromul Down este cea mai frecventă tulburare cromozomială. Cariotipul pacienților în 94% este format din 47 de cromozomi din cauza trisomiei pe cromozomul 21. În aproximativ 4% din cazuri, există o translocare a cromozomului 21 suplimentar la al 14-lea sau al 22-lea, numărul total de cromozomi este de 46. Boala se caracterizează printr-o întârziere bruscă și afectare fizică și fizică. dezvoltare mentală copil. Astfel de copii sunt subdimensionați, încep să meargă și să vorbească târziu. Aspectul copilului este izbitor (forma caracteristică a capului cu un occiput înclinat, o punte a nasului lată și adânc înfundată, o incizie mongoloidă a ochilor, o gură deschisă, creșterea anormală a dinților, macroglosie, hipotensiune musculară cu slăbire). articulații, brahidactilie, în special degetul mic, o cută transversală în palmă etc.) și întârziere mintală severă, uneori până la o idioție completă. Încălcările sunt observate în toate sistemele și organele. Malformațiile sistemului nervos (în 67%), cardiovascular (64,7%) sunt deosebit de frecvente. De regulă, reacțiile imunității umorale și celulare sunt modificate, sistemul de reparare a ADN-ului deteriorat suferă. Asociată cu aceasta este o susceptibilitate crescută la infecție, un procent mai mare de dezvoltare a neoplasmelor maligne, în special a leucemiei. În cele mai multe cazuri, pacienții sunt infertili. Există însă cazuri de naștere de copii de către o femeie bolnavă, unii dintre ei suferă de aceeași boală.
A doua cea mai frecventă patologie (1:5000-7000 de nașteri) datorată modificării numărului de autozomi este sindromul Patau (trisomia 13). Sindromul se caracterizează prin malformații severe ale creierului și feței (defecte ale structurii oaselor creierului și ale craniului facial, creierului, ochilor; microcefalie, buză despicată și palat), polidactilie (mai des - hexodactilie), defecte în sept inimii, rotația nearticulată a intestinului, boală polichistică de rinichi, defecte de dezvoltare ale altor organe. 90% dintre copiii născuți cu această patologie mor în primul an de viață.
Locul al treilea (1:7000 de nașteri) în rândul polisemiei autozomilor este ocupat de trisomia 18 (sindromul Edwards). Principalele manifestări clinice ale bolii: numeroase defecte ale sistemului osos (patologia structurii părții faciale a craniului: micrognatie, epicantus, ptoză, hipertelorism), cardiovasculare (defecte ale septului interventricular, defecte ale valvelor arteră pulmonară, aortă), hipoplazie unghială, rinichi potcoavă, criptorhidie la băieți. 90% dintre pacienți mor în primul an de viață.
Bolile cromozomiale asociate cu nedisjuncția cromozomilor sexuali sunt mult mai frecvente. Variantele cunoscute ale polisomiei gonozomale sunt prezentate în tabel.
Tipuri de polisomii gonozomale întâlnite la nou-născuți
(după N.P. Bochkov, A.F. Zaharov, V.I. Ivanov, 1984)
După cum reiese din tabel, numărul copleșitor de polisimie pe cromozomii sexuali se încadrează în trisomia XXX, XXV, XVV.
Cu cromozomul X trisomie („superfemeie”), semnele clinice ale bolii sunt adesea absente sau minime. Boala este diagnosticată prin detectarea a două corpuri Barr în loc de unul și prin cariotipul 47,XXX. În alte cazuri, pacienții prezintă hipoplazie a ovarelor, uterului, infertilitate, diferite grade de dizabilitate mintală. O creștere a numărului de cromozomi X în cariotip crește manifestarea retardului mintal. Astfel de femei sunt mai predispuse decât în populația generală să sufere de schizofrenie.
Variantele de polisomie care implică cromozomii Y sunt mai numeroase și mai diverse. Cel mai frecvent dintre ele - sindromul Klinefelter - se datorează unei creșteri a numărului total de cromozomi până la 47 din cauza cromozomului X. Un om bolnav (prezența cromozomului Y domină cu orice număr de cromozomi X) este diferit înalt, tip feminin de structură scheletică, inerție și retard mintal. Dezechilibrul genetic începe de obicei să se manifeste în timpul pubertății, subdezvoltarea caracteristicilor sexuale masculine. Testiculele sunt reduse în dimensiune, există aspermie sau oligospermie, adesea ginecomastie. Un semn de diagnostic de încredere al sindromului este detectarea cromatinei sexuale în celulele organismului masculin. Sindromul supercline-felter (ХХХУ, doi corpi Barr) se caracterizează printr-o severitate mai mare a acestor semne, eșecul psihic atinge gradul de idioție.
Proprietarul cariotipului 47, HUU - „super-om” este diferit comportament impulsiv cu elemente pronunţate de agresivitate. Un număr mare de astfel de persoane se găsesc printre prizonieri.
Monosomia gonozomală este mult mai puțin frecventă decât polisomia și este limitată doar la monosomia X (sindromul Shereshevsky-Turner). Cariotipul este format din 45 de cromozomi, nu există cromatina sexuală. Pacienții (femeile) se caracterizează prin statură mică, gât scurt, pliuri cutanate laterale cervicale. Caracterizat prin edem limfatic al picioarelor, slaba dezvoltare a caracteristicilor sexuale, absența gonadelor, hipoplazie a uterului și trompelor uterine, amenoree primară. Astfel de femei sunt sterile. Abilitatea mintală, de regulă, nu suferă.
Nu au fost găsite cazuri de monosomie Y. Aparent, absența cromozomului X este incompatibilă cu viața, iar indivizii de tip „OU” mor în stadiile incipiente ale embriogenezei.
Bolile cromozomiale cauzate de modificările structurale ale cromozomilor sunt mai puțin frecvente și, de regulă, duc la consecințe mai grave: avorturi spontane, prematuritate, naștere morta și mortalitate infantilă precoce.
In stadiile postimplantare nu au fost inregistrati fetusi cu trisomia cromozomilor 1 sau 19. Se presupune ca trisomia acestor cromozomi nu este deloc compatibila cu dezvoltarea postimplantare.10 blastomeri. Un caz de trisomie 1 mozaic în celulele citotrofoblaste a fost, de asemenea, înregistrat în studiile noastre. Aparent, în etapele ulterioare, astfel de embrioni fie mor, fie blastomerii cu un dezechilibru al acestor cromozomi sunt eliminați.
Trisomia 2 (Tc2) a fost descrisă numai în avorturi spontane. În același timp, se crede că Tc2 este caracteristic celulelor stromei mezenchimale a vilozităților coriale și este detectat numai pe preparatele de celule coriale cultivate. Cu toate acestea, am identificat un caz de Tc2 în citotrofoblast cu dezvoltarea sarcinii(Tabelul 5.5), iar literatura descrie cazuri de diagnostic prenatal și născuți vii a copiilor cu formă mozaică de Tc2.
Tc3 este una dintre cele mai frecvente caracteristici de trisomie a celulelor citotrofoblastice (8 cazuri în studiul nostru), iar proporția de celule trisomice poate varia de la descoperiri unice la forma completă.
Aparent, trisomiile cromozomilor din grupa B, precum și majoritatea cromozomilor din grupa C, sunt, de asemenea, letale și destul de rare chiar și în celulele corion. În studiile noastre, a fost înregistrat un caz al formei complete de trisomie 4, limitată la citotrofoblast.
atentie speciala merită cromozomii 7, 8 și 9, pentru care se remarcă o frecvență ușor crescută a trisomiilor corespunzătoare în materialul avorturilor spontane, comparativ cu ceilalți cromozomi din grupa C. Cazurile de Tc7, Tc8 și Tc9 detectate prenatal și la nou-născuți indică un efect subletal al unui exces de material genetic al acestor cromozomi. Prin urmare, prezența chiar și a unei forme de mozaic a acestor trisomii în celulele corionului necesită studiul cariotipului fetal. Se știe că Tc7 este una dintre trisomiile caracteristice trofoblastului (19 cazuri în studiile noastre). Între timp, formele mozaic ale trisomiei 7 sunt descrise și în culturile de celule din lichidul amniotic, precum și în fibroblastele pielii la copii după naștere. Prin urmare, opinia că Tc7 este întotdeauna limitată la citotrofoblast trebuie corectată. Forme complete de trisomie limitate de placenta pentru cromozomii de grup C
Tabelul 5.5. Frecvența (%) și spectrul anomaliilor cromozomiale în diferite stadii de ontogeneză
|
Cromozom |
Date proprii (rezultatele diagnosticului prenatal) N = 7579 |
Date din literatură |
||||
|
Dezvoltare persistând gravidă ness |
Mozaicismul limitat la placenta |
eu oh oh H VQ nn O o N O § |
mort născut |
Jivoroj bani |
Prognoza viabil proprietăți |
|
|
i |
- |
0,01 |
- |
- |
- |
- |
|
2 |
- |
0,01 |
1,1 |
- |
- |
0 |
|
3 |
- |
0,11 |
0,3 |
- |
- |
0 |
|
4 |
- |
0,01 |
0,8 |
- |
- |
0 |
|
5 |
- |
- |
0,1 |
- |
- |
0 |
|
6 |
- |
- |
0,3 |
- |
- |
0 |
|
7 |
0,026 |
0,23 |
0,9 |
- |
- |
0 |
|
8 |
- |
0,08 |
0,8 |
- |
- |
0 |
|
9 |
- |
0,05 |
0,7 |
0,1 |
- |
0 |
|
10 |
- |
0,01 |
0,5 |
- |
- |
0 |
|
11 |
- |
- |
0,1 |
- |
- |
0 |
|
12 |
- |
- |
0,2 |
- |
- |
0 |
|
13 |
0,2 |
0,02 |
1,1 |
0,3 |
0,05 |
2,8 |
|
14 |
- |
- |
1,0 |
- |
- |
0 |
|
15 |
- |
0,03 |
1,7 |
- |
- |
0 |
|
16 |
- |
0,05 |
7,5 |
- |
- |
0 |
|
17 |
- |
- |
0,1 |
- |
- |
0 |
|
18 |
0,77 |
0,01 |
1,1 |
1,2 |
0,01 |
5,4 |
|
19 |
- |
- |
- |
- |
- |
0 |
|
20 |
- |
0,05 |
0,6 |
- |
- |
0 |
|
21 |
1,64 |
0,1 |
2,3 |
1,1 |
0,12 |
22,1 |
|
22 |
0,013 |
0,05 |
2,7 |
0,1 |
- |
0 |
|
Mozaic trisomie |
0,05 |
- |
1,1 |
0,5 |
0,02 |
9,0 |
|
Dubla trisomie |
- |
0,01 |
0,8 |
- |
- |
0 |
|
XXY |
0,19 |
- |
0,2 |
0,4 |
0,05 |
55,3 |
|
XXX |
0,09 |
- |
0,1 |
0,3 |
0,05 |
70,0 |
|
XYY |
0,05 |
- |
- |
- |
0,05 |
100,0 |
|
45,X |
0,43 |
0,4 |
8,6 |
0,25 |
lt; 0,01 |
0,3 |
|
Poliploidie |
0,25 |
0,01 |
9,8 |
0,6 |
- |
0 |
|
Structural |
0,12 |
0,01 |
2,0 |
0,4 |
0,6 |
62,0 |
(în special 6, 7 și 11, în care grupurile de gene imprimate sunt localizate), necesită un diagnostic precis al cariotipului fetal și excluderea disomiei uniparentale.
Trisomia 13 (sindromul Patau) este subletal printre trisomiile cromozomilor de grup D (13, 14, 15). Este interesant de observat că formele complete ale acestei trisomie sunt mai frecvente decât cele din mozaic, inclusiv cele limitate la placentă. Trisomia letală 14 și 15, identificată în trofoblast, merită atenție în ceea ce privește disomia uniparentală la făt. Prin urmare, dacă există celule cu trisomie a oricăruia dintre cromozomii grupului D în proba de corion, este necesară cariotiparea fătului de către limfocitele din sângele din cordonul ombilical.
Tc16 este una dintre cele mai frecvente aberații numerice pe primele etape dezvoltare (dintre avorturile spontane, frecvența acestuia este de 7,5%). Interesant, în proba noastră au fost identificate doar un caz de trisomie 16 completă și două cazuri cu celule trisomice unice în citotrofoblastul placentar. Din păcate, cariotipul fetal nu a fost studiat în niciun caz. Cu toate acestea, cazurile de Tc16 în celulele lichidului amniotic descrise în literatură sugerează că, cel puțin, mozaicurile cu o astfel de tulburare de cariotip se pot dezvolta înainte de al doilea trimestru de sarcină.
Cazurile de Tc17 nu au fost identificate în studiile noastre. Într-o variantă de mozaic, ele sunt descrise în amniocitele din al doilea trimestru, dar frecvența lor este scăzută.
Tc18 (sindromul Edwards) ca mutație subletală apare în toate etapele dezvoltării intrauterine. Ca și alte trisomii subletale frecvente, Tc18 este reprezentată în principal de forme complete și mult mai rar de cele mozaic. În studiul nostru, Tc18 a fost limitat la placentă într-un singur caz, în timp ce alți autori notează o frecvență ridicată a Tc18 în corion.
Tc20 a fost mult timp considerat letal în stadiile embrionare timpurii. În prezent, cazuri de mozaic de Tc20 au fost detectate prenatal în diferite etape ale sarcinii și la copii. Cu toate acestea, complexul de defecte în Tc20 nu a fost identificat ca un sindrom specific. Interesant este că Tc20 este de obicei limitat la celulele țesuturilor extraembrionare, în timp ce la făt este prezent doar în celulele anumitor organe (rinichi, rect, esofag). Toate cele 4 cazuri de Tc20 complet și mozaic din studiul nostru s-au limitat la celule trofoblastice.
Conform numeroaselor observații, pentru Tc21 (sindromul Down), forma completă este caracteristică. În studiile noastre, mozaic Tc21 cu o linie diploidă dominantă în citotrofoblast a fost găsit în 4 cazuri. În niciunul dintre ele, diagnosticul a fost confirmat prin studiul limfocitelor din sângele din cordonul ombilical fetal sau al sângelui periferic neonatal. Cu toate acestea, credem că toate cazurile de mozaic Tc21 în citotrofoblast necesită studii suplimentare asupra altor celule (amniocite, limfocite din sângele din cordonul ombilical), deoarece prognoza de viabilitate la fetușii cu trisomie 21, spre deosebire de alte trisomii subletale, este de obicei favorabilă (22.1). %) (Tabelul 5.4).
Se știe că Tc22 există ca un sindrom Tc22 independent, adică este subletal. Formular complet Tc22 a fost înregistrat de noi în citotrofoblast doar într-un caz; în alte trei era reprezentată printr-o variantă mozaică. Cromozomul uman 7 păianjen, organele cromozomului 7 uman
al 7-lea cromozom uman unul dintre cei 23 de cromozomi umani. Cromozomul conține mai mult de 158 de milioane de perechi de baze, ceea ce reprezintă între 5 și 5,5% din materialul total ADN al unei celule umane. în prezent se crede că pe cromozomul 7 există de la 1000 la 1400 de gene.
Cromozomul 7 conține grupul A de gene homeobox.
- 1 Genele
- 1.1 Umăr p
- 1.2 Umăr q
- 2 Boli și tulburări
- 2.1 Boli cromozomiale
- 3 note
Genele
Unele dintre genele situate pe cromozomul 7 sunt enumerate mai jos:
Umăr p
- ABCA13 este al 13-lea membru al subfamiliei A a proteinelor casete care leagă ATP;
- AQP1 - Aquaporin 1;
- C1GALT1 - glicoziltransferaza;
- CBX3 - omologul cromobox 3;
- CCM2 - malformație cavernoasă cerebrală 2;
- DFNA5 - surditate, autosomal dominant tip 5;
- GARS, glicil-ARNt sintetaza;
- IL6 - interleukina 6;
- ITGB8 este o glicoproteină din superfamilia integrinelor (β8);
- NOD1 - receptor asemănător Nod al subfamiliei NOD;
- PMS2 - PMS2 engleză segregarea postmeiotică a crescut 2 (S. cerevisiae).
umăr q
- ABCB1 - glicoproteină P;
- ASL - argininosuccinat liază;
- CAV1 - caveolina 1;
- CCL24 - Chemokine (motiv C-C) ligand 24 (scya24);
- CCL26 - Chemokine (motiv C-C) ligand 26 (scya26);
- CD36;
- CDK5 - kinaza 5 dependentă de ciclină;
- CGRP-RCP - proteină componentă a receptorului peptidic legat de gena calcitoninei;
- CFTR - Regulator transmembranar al fibrozei chistice, casetă de legare a ATP (subfamilia C, membru 7);
- CLCN1 - canalul de clorură 1;
- CNTNAP2 - genă asociată cu autismul;
- COL1A2 - colagen, tip I, alfa 2;
- CYLN2 - linker citoplasmatic 2;
- DLD - dihidrolipoamidă dehidrogenază (componenta E3 a complexului de piruvat dehidrogenază, complex 2-oxo-glutarat, complex cetoacid dehidrogenază cu lanț ramificat);
- ELN - elastina (stenoza aortica supravalvulara, sindromul Williams-Beuren);
- FOXP2 - Cutie cu furcă proteină 2;
- GTF2I - factorul general de transcripție II, i;
- GTF2IRD1 - domeniu repetat GTF2I care conține 1;
- GUSB - beta-glucuronidază;
- HSPB1 - soc termic 27kDa proteina 1;
- KCNH2 - canal voltaj de potasiu, subfamilia H (înrudit cu ouă), membru 2;
- KRIT1 - KRIT1, care conține repetiții anchirine;
- LIMK1 - kinaza domeniului LIM 1;
- NOS3 - oxid nitric sintază endotelial
- p47 phox sau NCF1 - factor neutrofil oxidază de 47 kDa/factor citosolic neutrofil 1;
- PIK3CG - subunitatea catalitică y a fosfatidilinozitol-4,5-bisfosfat-3-kinazei (PI3K gamma, p110γ);
- RELN - reelin;
- SBDS - sindromul Shwachman-Bodian-Diamond;
- SH2B2 - proteină adaptoare;
- SLC25A13 - purtător solvent familia 25, membru 13 (citrină);
- SLC26A4 - rezolvă familia purtătoare 26, membru 4;
- SRI - sorcin;
- TAS2R16 - receptor gustativ, tip 2, membru 16;
- TFR2 - receptor 2 al transferinei;
- TPST1 - tirozil protein sulfotransferaza 1;
- VGF - „factor de creștere a nervilor inductibil”.
Boli și tulburări
Mai jos sunt enumerate câteva dintre bolile asociate cu genele de pe al 7-lea cromozom, precum și genele ale căror defecte provoacă aceste boli:
- argininosuccinic aciduria (în engleză argininosuccinic aciduria) - ASL;
- surditate nonsindromică autosomal dominant tip 5 și autosomal recesiv tip 4 - DFNA5 și SLC26A4;
- boala urinei cu sirop de arțar - DLD;
- Boala Charcot-Marie-Tooth tipurile 2B și 2F - GARS și HSPB1;
- absența bilaterală congenitală a canalelor deferente - CFTR;
- hemocromatoză tip 3 (hemocromatoză engleză, tip 3) - TFR2;
- atrofie musculară spinală distală tip 5 - GARS;
- angiom cavernos (malformație cavernosă cerebrală engleză) - CCM2;
- sindrom mielodisplazic;
- fibroza chistica - CFTR;
- mucopolizaharidoza tip VII, sau sindromul Sly - GUSB;
- cancer colorectal ereditar nonpolipoz - PMS2;
- osteogeneza imperfectă tipurile I, II, III şi IV - COL1A2;
- diabet zaharat tip adult la tineri tip 2 - GCK;
- Sindromul Williams - ASL, BAZ1B, BCL7B, CLDN3, CLDN4, CLIP2, EIF4H, ELN, FZD9, FKBP6, GTF2I, GTF2IRD1, HIP1, KCTD7, LAT2, LIMK1, MDH2, NCF1, NSUN5, POR, RCAFC20, TBL2, STX1 , TRIM73, TRIM74, WBSCR14, WBSCR18, WBSCR21, WBSCR22, WBSCR23, WBSCR24, WBSCR27 și WBSCR28;
- sindromul Norman-Roberts - RELN;
- sindromul Pendred - SLC26A4;
- sindromul Romano-Ward - KCNH2;
- sindromul Shwachman-Diamond (SBDS);
- sindromul Ehlers-Danlos cu artrocalazie tip 7B - COL1A2;
- boală granulomatoasă cronică datorată deficienței factorului citosolic neutrofil 1 - NCF1
- citrulinemie (citrulinemia engleză) tip II - SLC25A13;
- schizofrenie - KCNH2, ABCA13
Boli cromozomiale
Unele tulburări sunt cauzate de modificări ale structurii sau numărului de copii ale cromozomului 7:
- Sindromul Williams - o ștergere a unei secțiuni a brațului lung al cromozomului la poziția 7q11.23, care conține mai mult de 20 de gene; pierderea unora dintre aceste gene este asociată cu trasaturi caracteristice tulburări, dar majoritatea genelor site-ului șterse nu au fost încă asociate cu simptome;
- creștere și dezvoltare întârziată, retard mental, modificări caracteristice ale trăsăturilor faciale, anomalii ale scheletului, vorbire lentă și alte probleme medicale - o copie suplimentară a unei părți a cromozomului (trisomie parțială) sau absența unei părți a cromozomului (monosomie parțială), uneori o ștergere sau o duplicare a unei părți a cromozomului și a cromozomului inel de apariție (cromozom inel în engleză).
Note
- Harta cromozomului uman 7. Baza de date VEGA (Vertebrate Genome Annotation). Institutul Wellcome Trust Sanger. - O hartă a cromozomului și a parametrilor săi principali: dimensiunea, numărul de gene etc. Consultat la 8 septembrie 2009. Arhivat din original la 1 aprilie 2012.
- Kratz C. P., Emerling B. M., Bonifas J., Wang W., Green E. D., Le Beau M. M., Shannon K. M. Structura genomică a genei PIK3CG pe banda cromozomală 7q22 și evaluarea ca supresor de tumori mieloide candidat // Sânge. - 2002. - T. 99, nr. 1. - S. 372-374. - PMID 11756194.
Cariotipul celulelor măduvei osoase la pacienți Cu(MDS) a fost studiat intens în ultimii 10-15 ani. Clonele anormale sunt identificate înainte de tratament la 30-50% dintre pacienți, în unele rapoarte fiind date rate mai mari - 60-75%.
Detectarea clonelor celulare cu anomalii cariotipîn sindromul mielodisplazic (MDS) are o importantă teoretică şi semnificație clinică, deoarece indică faptul că acest grup de boli aparține neoplasmelor.
Modificările citogenetice sunt foarte variat, spectrul lor este apropiat de spectrul anomaliilor cromozomiale observate în leucemia acută non-limfoblastică, în special în cele secundare.
Cel mai caracteristic monosomia 5 și 7, precum și deleții ale brațului lung al acestor cromozomi, apariția unui cromozom 8 suplimentar și ștergerea brațului lung al cromozomului 20.
Se constată că frecvenţa detectare clonele de celule aneuploide crește odată cu progresia bolii: în stadii relativ incipiente este de 20-30%, cu apariția semnelor inițiale de transformare în leucemie acută- până la 40-60%, cu transformare în leucemie mieloidă acută - 80-90%.
Translocatii specifice primarului leucemii acute non-limfoblastice sunt rare în mielodisplazie. Există rapoarte de translocații repetate t(3;3)(q21;q26), t(8;21)(q22;q22) și t(3;21)(q26;q22). Exemple de rearanjamente ale brațului lung al cromozomului 3 sunt prezentate în figură.
Frecvența (în procente) a anomaliilor cariotipului caracteristice în diferite sindroame mielodisplazice
Anomalii cromozomiale majore caracteristice mielodisplaziilor:
-7 sau 7q-
-5 sau 5q-
t(1;7)(q10;p10)
del(12)(p12-p13)
t(2;ll)(p13;q23)
del(13) (necesar să includă 3q14)
t(6;9)(p23;q34)
del(20)(q11ql3)
+8
t(1;3)(p36;q21)
Listată anomalii cromozomiale observat la diferite forme mielodisplazie, dar frecvența lor este oarecum diferită.
Cea mai mare experiență cercetători indică faptul că există o corelație între caracteristicile cariotipului și speranța de viață a pacienților cu sindrom mielodisplazic (MDS). Prognosticul este considerat relativ favorabil dacă sunt detectate clone celulare cu o singură rearanjare 5q- sau 20q; totodată, în orice variantă de sindrom mielodisplazic, depistarea unei clone cu anomalii cromozomiale multiple este extrem de nefavorabilă.
Să ne oprim Mai mult asupra tulburărilor de cariotip individuale caracteristice sindromului mielodisplazic (MDS).
Sindromul 5q- anemie sideroblastică refractară la pacienții vârstnici, în principal femei. În noua clasificare a OMS, acest sindrom se distinge ca varianta independenta sindrom mielodisplazic (MDS). Caracterizată prin anemie macrocitară, rezistentă la tratament, în măduva osoasă - semne de mielodisplazie a eritrocitelor și megacariocitelor. Numărul de trombocite este normal sau crescut, în măduva osoasă se observă hiperplazia micromegacariocitelor hipolobulare. Cursul clinic este relativ lent. Transformarea în leucemie acută are loc în aproximativ 10% din cazuri. Sindromul a fost descris pentru prima dată de van den Berghe și colab. în 1974-1985.
Ștergeri brațul lung al cromozomului 5 observată în alte boli hematologice.
Se presupune că ștergerea site-ului conţine una sau mai multe gene supresoare. În această direcție se desfășoară cercetări intense. Pana acum nu s-a confirmat rol importantîn patogenia anemiei refractare niciunul dintre candidaţii studiati pentru rolul genei supresoare.
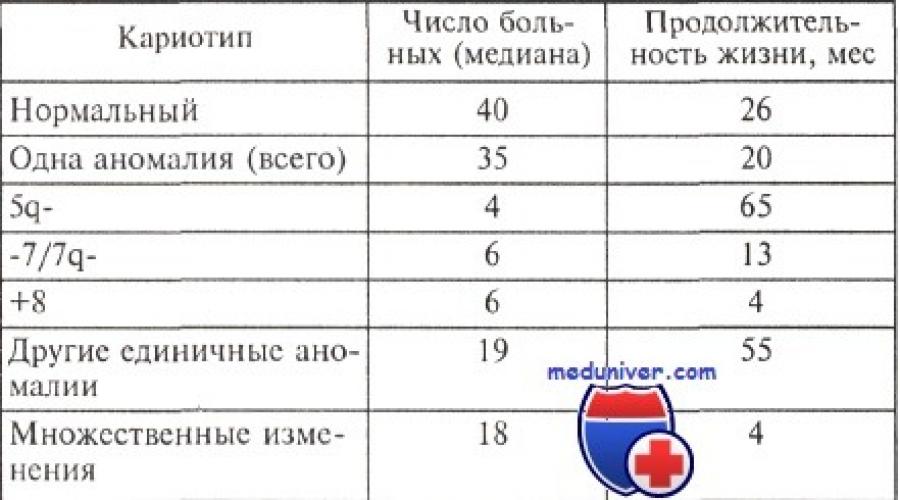
Speranța de viață a pacienților cu sindrom mielodisplazic cu diferite modificări ale cariotipului

Sindromul monozomic al cromozomului 7 apare predominant la băieții sub 4 ani. Splenomegalia este caracteristică, se observă adesea leucocitoză cu monocitoză, trombocitopenie, anemie. Prognosticul este prost.
După cum sa menționat, pierderea unuia dintre perechea cromozomului 7(monozomia 7) se observă într-o mare varietate de hemoblastoze, inclusiv leucemia acută non-limfoblastică, și este de obicei asociată cu un prognostic prost.
Deleții ale brațului scurt al cromozomului 17 (17p-) sunt de obicei incluse în modificări complexe de cariotip. De regulă, 17p- este asociat cu două sau mai multe anomalii cromozomiale și are o valoare prognostică nefavorabilă.
În 75% din cazuri în prezență marker 17r- există un fel de disgranulocitopoieză sub formă de nuclei hipolobulari pseudo-pelgerieni și vacuolizarea citoplasmei. Acest marker se găsește nu numai în mielodisplazie, ci și într-o mare varietate de neoplasme maligne, inclusiv tumori solide, prezența sa fiind un semn de prognostic slab.
În 1997, materialele internaționale întâlniri dedicat diagnosticului și prognosticului sindromului mielodisplazic. Pe baza unei evaluări retrospective a duratei bolii înainte de tranziția la leucemie acută și a speranței totale de viață a pacienților, rezultatul analizei citogenetice a fost considerat cel mai important semn de prognostic. Lotul cu prognostic favorabil include cazurile cu anomalii cromozomiale unice: -Y, 5q- si 20q-. curs nefavorabil observate în tulburări multiple (complexe) (trei sau mai multe rearanjamente ale cariotipului) și modificări ale cromozomului 7 (deleții ale brațului lung, monosomie).
Alte anomalii au determinat prognosticul „interimar”. Durata medie a bolii înainte de trecerea la leucemie acută a fost de 9,4 per grup; 0,4 și, respectiv, 1,1-3,3 ani. Aceste date sunt folosite pentru a evalua eficacitatea noilor scheme de tratament a mielodisplaziei și s-au dovedit a fi unul dintre cele mai bune sisteme de predicție pentru sindromul mielodisplazic.
Metoda poate avea o mare valoare diagnostică. PEŞTEîn cazurile în care un studiu citogenetic standard nu este informativ sau se găsesc doar celule unice cu tulburare de cariotip, care, după criterii formale, nu pot fi considerate o clonă. Un grup de sonde FISH este în curs de dezvoltare pentru a diagnostica cele mai caracteristice anomalii cromozomiale în sindromul mielodisplazic.
Încercări evidențiază caracteristicile citogenetice fiecare dintre subunitățile clinice și morfologice incluse în grupul eterogen general de sindroame mielodisplazice nu a avut succes. În același timp, CMML, considerată ca o boală mieloproliferativă cu semne morfologice de mielodisplazie, este adesea asociată cu o anomalie cromozomială specifică t(5;12)(q33;p13), totuși, în majoritatea cazurilor de CMML, această anomalie cromozomială este nu a fost detectat.