Апоптоз генетически запрограммированная гибель клетки. Апоптоз
Читайте также
При реализации апоптоза условно можно выделить четыре стадии.
Инициация -> Программирование -> Реализация программы -> Удаление погибшей клетки
Стадии апоптоза Стадия инициации
На этой стадии информационные сигналы рецептируются клеткой . Патогенный агент либо сам является сигналом, либо обусловливает генерацию сигнала в клетке и его проведение к внутриклеточным регулятор-ным структурам и молекулам.
Инициирующие апоптоз стимулы могут быть трансмембранными или внутриклеточными.
Трансмембранные сигналы подразделяют на отрицательные, положительные и смешанные.
- Отрицательные сигналы : отсутствие или прекращение воздействия на клетку факторов роста, цитокинов, регулирующих деление и созревание клетки, а также гормонов, контролирующих развитие клеток. В норме действие названных выше групп БАВ на мембранные рецепторы обеспечивает подавление программы гибели клеток и нормальную их жизнедеятельность. Напротив, их отсутствие или снижение эффектов «освобождает» программу апоптоза. Так, для нормальной жизнедеятельности ряда нейронов необходимо постоянное наличие нейротрофических факторов. Их устранение или снижение эффектов на нервные клетки может привести к включению программы смерти нейрона. - Положительные сигналы в итоге генерируют запуск программы апоптоза . Так, связывание ФИО (FasL) с его мембранным рецептором CD95 (Fas) активирует программу смерти клетки. - Смешанные сигналы являются комбинацией воздействий сигналов первой и второй групп. Так, апоптозу подвергаются лимфоциты, простимулированные митогеном, но не проконтактировавшие с чужеродным Аг. Погибают и те лимфоциты, на которые воздействовал Аг, но не получившие других сигналов, например митогенного или от HLA.
Среди внутриклеточных стимулов апоптоза зарегистрированы избыток Н+, свободные радикалы липидов и других веществ, повышенная температура, внутриклеточные вирусы и гормоны, реализующие свой эффект через ядерные рецепторы (например, глюкокортикоиды).
Апоптоз: стадия инициации.
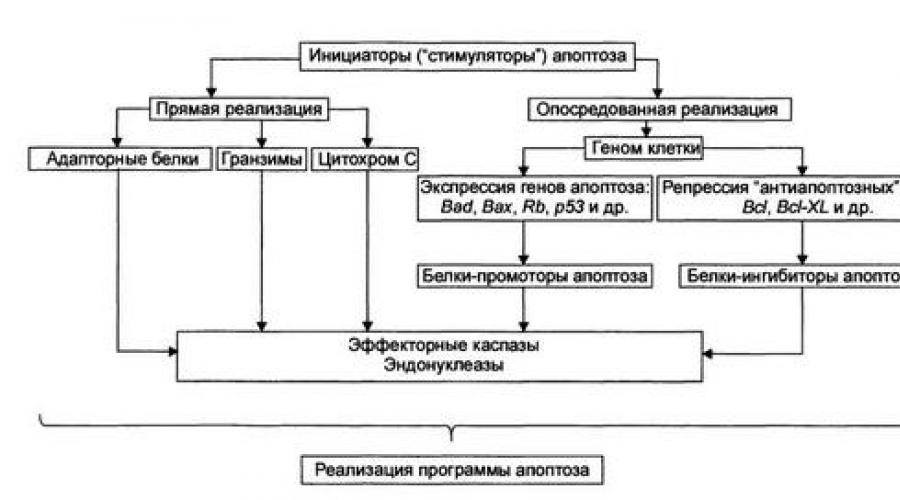
Стадия программирования
Стадия программирования (контроля и интеграции процессов апоптоза) представлена на рисунке.
На этой стадии специализированные белки либо реализуют сигнал к апоптозу путём активации исполнительной программы (её эффекторами являются цистеиновые протеазы - каспазы и эндонуклеазы), либо блокируют потенциально летальный сигнал.
Выделяют два (не исключающих друг друга) варианта реализации стадий программирования : 1) путём прямой активации эффекторных каспаз и эндонуклеаз (минуя геном клетки) и 2) опосредованной через геном передачи сигнала на эффекторные каспазы и эндонуклеазы.
Прямая передача сигнала осуществляется через адапторные белки, гранзимы и цитохром С.
Адапторные белки . В качестве адапторного белка выступает, например, каспаза-8. Так реализуют своё действие цитокины Т-лимфоцитов-киллеров в отношении чужеродных клеток, ФНО и другие лиганды CD95.
Цитохром С . Выделяясь из митохондрий, цитохром С вместе с белком Apaf-1 и каспазой-9 формирует комплекс активации (апоптосому) эффекторных каспаз. Каспаза-8 и каспаза-9 активируют эффекторные каспазы (например, каспазу-3), которые участвуют в протеолизе белков.
Гранзимы . Эти протеазы выделяют цитотоксические Т-лимфоциты, протеазы проникают в клетки-мишени через цитоплазматические поры, предварительно сформированные перфоринами. Гранзимы активируют аспартатспецифи-ческие цистеиновые протеазы клетки-мишени, подвергающейся апоптозу.
Прямая передача сигнала наблюдается обычно в безъядерных клетках, например в эритроцитах.

Апоптоз: стадия программирования.
Опосредованная передача сигнала подразумевает репрессию генов, кодирующих ингибиторы апоптоза, и активацию генов, кодирующих промоторы апоптоза.
Белки-ингибиторы апоптоза (например, продукты экспрессии антиапоптозных генов Bcl-2, Bcl-XL) блокируют апоптоз (например, путём уменьшения проницаемости мембран митохондрий, тем самым уменьшая вероятность выхода в цитозоль одного из пусковых факторов апоптоза - цитохрома С).
Белки-промоторы апоптоза (например, белки, синтез которых контролируется генами Bad, Box, антионкогенами Rb или /т53) активируют эффекторные кас-пазы и эндонуклеазы.
Определение апоптоза. Апоптоз – феномен наследственно запрограммированной смерти клеток. Каждая клетка при своем рождении как бы запрограммирована на самоуничтожение. Условие ее жизни – блокирование этой суицидальной программы.
Апоптоз реализуется для клеток:
Старых, отживших свой срок;
Клеток с нарушениями дифференцировки;
Клеток с нарушениями генетического аппарата;
Клеток, пораженных вирусами.
Морфологические признаки апоптоза.
Сморщивание клетки;
Конденсация и фрагментация ядра;
Разрушение цитоскелета;
Буллезное выпячивание клеточной мембраны.
Особенность апоптоза – апоптоз не вызывает воспаления в окружающих тканях.Причина - сохранность мембраны и → изоляция повреждающих факторов цитоплазмы до полного завершения процесса (О 2 - , Н 2 О 2 , лизосомальные ферменты). Эта особенность – важная позитивная черта апоптоза, в отличие от некроза. При некрозе мембрана повреждается (или разрывается) сразу же. Поэтому при некрозе содержимое цитоплазмы высвобождается (О 2 - , Н 2 О 2 , лизосомальные ферменты). Возникает повреждение соседних клеток и воспалительный процесс. Важная черта апоптоза - удаление умирающих клеток происходит без развития воспаления.
Процесс апоптоза - может быть разделен на 2 (две) фазы:
1. Формирование и проведение апоптических сигналов – фаза принятия решения.
2. Демонтаж клеточных структур – эффекторная фаза.
1-я фаза – принятия решения (=формирование и принятие апоптических сигналов). Это фаза принятия стимулов для апоптоза. В зависимости от характера стимулов, может быть 2 (два) типа сигнальных путей:
1) повреждение ДНК в результате радиации, действия токсических агентов, глюкокортикоидов и т.д.
2) активация рецепторов «региона клеточной смерти» . Рецепторы «региона клеточной смерти» - это группа рецепторов на мембранах любых клеток, которые воспринимают проапоптические стимулы. Если количество и активность таких рецепторов увеличивается, то увеличивается количество апоптически гибнущих клеток. К рецепторам «региона клеточной смерти» относятся: а) TNF-R (связывается с фактором некроза опухолей и активирует апоптоз); б) Fas-R (к); в) CD45-R (связывается с антителами и активирует апоптоз).
В зависимости от типа сигнала, существует 2 (два) основных способа апоптоза: а) в результате повреждения ДНК;
б) в результате самостоятельной активации рецепторов «региона клеточной смерти» без повреждения ДНК.
2-я фаза – эффекторная (=демонтаж клеточных структур. Основные фигуранты эффекторной фазы:
Цистеиновые протеазы (каспазы);
Эндонуклеазы;
Сериновые и лизосомальные протеазы;
Протеазы, активированные Ca ++ (кальпейн)
Но! Среди них основные эффекторы демонтажа клеточных структур – каспазы.
Классификация каспаз - 3 (три) группы:
Эффекторные каспазы - каспазы 3, 6, 7.
Индукторы активации эффекторных каспаз – каспазы 2, 8, 9, 10. = активаторы цитокинов – каспазы 1, 4, 5, 13.
Эффекторные каспазы – каспазы 3, 6, 7. Это непосредственные исполнители апоптоза. Эти каспазы находятся в клетке в неактивном состоянии. Активированные эффекторные каспазы начинают цепь протеолитических событий, целью которых является «демонтаж» клетки. Их активируют индукторы активации эффекторных каспаз.
Индукторы активации эффекторных каспаз – каспазы 2, 8, 9, 10. Основные индукторы – каспазы 8 и 9 . Они активируют эффекторные каспазы. Механизм – расщепление аспарагиновых оснований с последующей димеризацией активных субъединиц. Эти каспазы при обычном состоянии в клетках неактивны, существуют в форме прокаспаз.
Активация тех или иных индукторов зависит от типа сигнального пути:
1. При повреждении ДНК задействован сигнальный путь № 1, активируется каспаза № 9.
2. При активации рецепторов клеточной смерти задействован сигнальный путь № 2, активируется каспаза № 8.
Сигнальный путь № 1 (связан с повреждением ДНК)
Повреждение ДНК
Активация гена р53 и продукция соответствующего белка
Активация проапоптических генов семейства BCL-2 (BAX и BID)
Образование белков этих генов
Активация каспазы 9
Активация каспазы 3
Сигнальный путь № 2
(связан с активацией «региона клеточной смерти»)
Лиганд + рецепторы «региона клеточной смерти»
Активация каспазы № 8
Независимая активация каспазы № 3
Активация других каспаз и протеаз
Регуляция апоптоза. Исследования последних лет привели к созданию модели апоптоза. По этой модели каждая клетка при своем рождении запрограммирована на самоуничтожение. Следовательно, условием ее жизни является блокирование этой суицидальной программы. Основная задача регуляции апоптоза – держать эффекторные каспазы в неактивном состоянии, но быстро переводить их в активную форму в ответ на минимальное действие соответствующих индукторов.
Отсюда, понятие ингибиторов и активаторов апоптоза.
Ингибиторы апоптоза (=антиапоптические факторы). К наиболее серьезным ингибиторам апоптоза относятся ростовые факторы. Другие: нейтральные аминокислоты, цинк, эстрогены, андрогены, некоторые белки.
Пример: Белки семейства IAP – подавляют активность каспаз 3 и 9. Запомнить: один из этих белков (Survin) обнаружен в опухолевых клетках. С ним связывают резистентность опухолевых клеток к химиотерапии
Активаторы апоптоза (=проапоптические факторы). Это проапоптические гены и их продукция: а) гены семейства BCL-2 (BAX и BID); б) гены Rb и P53 (запускают апоптоз, если клетка задержана механизмом checkpoint.
Резюме. Патогенез многих заболеваний, в том числе и опухолевых, связан со снижением способности клеток подвергаться апоптозу. Отсюда накопление поврежденных клеток и формирование опухоли.
ПАТОФИЗИОЛОГИЯ КЛЕТОЧНОГО ДЕЛЕНИЯ
Основное отличие деления здоровой и опухолевой клетки:
Деление здоровой клетки регулируется паракринным и эндокринным способом. Клетка подчиняется этим сигналам и делится только в том случае, если организм нуждается в образовании новых клеток данного вида.
Деление опухолевой клетки регулируется аутокринным способом. Опухолевая клетка сама образует митогенные стимуляторы и сама же делится под их влиянием. Она не отвечает на паракринные и эндокринные стимулы.
Существует 2(два) механизма опухолевой трансформации клеток:
1. Активация онкогенов.
2. Инактивация генов-супрессоров.
АКТИВАЦИЯ ОНКОГЕНОВ
Прежде всего 2 (два) главных понятия: = протоонкогены;
Онкогены.
Протоонкогены – это нормальные, неповрежденные гены, которые контролируют деление здоровой клетки.
К протоонкогенам относятся гены, контролирующие образование и работу:
1. Ростовых факторов.
2. Мембранных рецепторов к ростовым факторам, например тирозинкиназных рецепторов.
3. Ras-белков.
4. MAP-киназ, участниц МАР-киназного каскада.
5. Транскрипционных факторов AP-1.
Онкогены – поврежденные протоонкогены. Процесс повреждения протоонкогена и трансформация его в онкоген называется активация онкогена.
Механизмы активации онкогена.
1. Включение (вставка) промотора. Промотор – это участок ДНК, с которым связывается РНК-полимераза протоонкогена. Необходимое условие – промотор должен находится в непосредственной близости с протоонкогеном. Отсюда варианты: а) промотор - ДНК-копия онкорнавирусов; б) «прыгающие гены» - участки ДНК, способные перемещаться и встраиваться в разные участки генома клетки.
2. Амплификация – увеличение числа протоонкогенов или появление копий протоонкогенов. Протоонкогены в норме обладают небольшой активностью. При увеличении числа или появлении копий их общая активность значительно возрастает и это может привести к опухолевой трансформации клетки.
3. Транслокация протоонкогенов. Это перемещение протоонкогена в локус с функционирующим промотором.
4. Мутации протоонкогенов.
Продукция онкогенов. Онкогены образуют свои белки. Эти белки называются «онкобелки».
Синтез онкобелков называется «экспрессия активных клеточных онкогенов».
Онкобелки – в основе своей есть аналоги белков протоонкогенов: ростовых факторов, Ras-белков, МАР-киназ, транскрипционных факторов. Но есть количественные и качественные отличия онкогенов от белков протоонкогенов.
Отличия онкобелков от нормальной продукции протоонкогенов:
1. Увеличение синтеза онкобелков по сравнению с синтезом белков протоонкогенов.
2. Онкобелки имеют структурные отличия от белков протоонкогенов.
Механизм действия онкобелков.
1. Онкобелки соединяются с рецепторами для факторов роста и образуют комплексы, постоянно генерирующие сигналы к делению клетки.
2. Онкобелки повышают чувствительность рецепторов к факторам роста или понижают чувствительность к ингибиторам роста.
3. Онкобелки могут сами действовать как факторы роста.
ИНАКТИВАЦИЯ ГЕНОВ-СУПРЕССОРОВ
Гены-супрессоры: Rb и р53.
Их продукция – соответствующие белки.
Инактивация генов-супрессоров (наследственное или приобретенное) ведет к пропуску в митоз клеток с поврежденной ДНК, размножению и накоплению этих клеток. Это – возможная причина формирования опухоли.
ОПУХОЛЕВЫЙ РОСТ: ОПРЕДЕЛЕНИЕ, ПРИЧИНЫ УВЕЛИЧЕНИЯ КОЛИЧЕСТВА ЗЛОКАЧЕСТВЕННЫХ ЗАБОЛЕВАНИЙ
Опухоль – патологическое разрастание, отличающееся от других патологических разрастаний наследственно закрепленной способностью к неограниченному неконтролируемому росту.
Другие патологические разрастания – гиперплазия, гипертрофия, регенерация после повреждения.
Причины увеличения количества злокачественных заболеваний среди населения:
1. Увеличение продолжительности жизни.
2. Улучшение качества диагностики → увеличение выявляемости онкологических заболеваний.
3. Ухудшение экологической обстановки, увеличение содержания канцерогенных факторов в окружающей среде.
ДОБРОКАЧЕСТВЕННЫЕ И ЗЛОКАЧЕСТВЕННЫЕ ОПУХОЛИ
Единой классификации опухолей до сих пор не создано. Причина:
1. Большое разнообразие признаков, характерных для различных опухолей.
2. Недостаточность знания их этиологии и патогенеза.
В основе современных классификаций - главные морфологические и клинические признаки опухолей.
На основе клинической характеристики все опухоли делят на доброкачественные и злокачественные.
Доброкачественные опухоли:
1. Клетки опухоли морфологически идентичны или похожи на нормальные клетки-предшественники.
2. Степень дифференцировки опухолевых клеток – достаточно высокая.
3. Скорость роста – медленная, в течение многих лет.
4. Характер роста – экспансивный, т.е. во время роста опухоли соседние ткани раздвигаются, иногда сдавливаются, но обычно не повреждаются.
5. Отграниченность от окружающих тканей – четкая.
6. Способность к метастазированию – отсутствует.
7. Отсутствие выраженного неблагоприятного воздействия на организм. Исключение: опухоли, расположенные вблизи жизненно важных центров. Пример: опухоль головного мозга, сдавливающая нервные центры.
Злокачественные опухоли.
1. Клетки опухоли морфологически отличаются от нормальной клетки-предшественницы (часто до неузнаваемости).
2. Степень дифференцировки опухолевых клеток – низкая.
3. Скорость роста – быстрая.
4. Характер роста – инвазивный, т.е. опухоль прорастает в соседние структуры. Способствующие факторы:
Приобретение опухолевыми клетками способности отшнуровываться от опухолевого узла и активно перемещаться;
Способность опухолевых клеток продуцировать «канцероагрессины». Это белки, которые проникают в окружающие нормальные ткани и стимулируют хемотаксис для опухолевых клеток.
Уменьшение сил клеточной адгезии. Это облегчает отшнуровку опухолевых клеток от первичного узла и их последующее движение.
Уменьшение контактного торможения.
5. Отграниченность от окружающих тканей – нет.
6. Способность к метастазированию – выражена.
7. Воздействие на организм – неблагоприятное, генерализованное.
Явления запрограммированной клеточной гибели известны уже более 100 лет, но оставались «в тени» некробиотических процессов, которые на протяжении десятилетий изучались намного более активно, чем программируемая гибель. Этот вид клеточной гибели представляет собой важнейший интегральный компонент эмбриогенеза, морфогенеза и роста тканей, а также гормонозависимой инволюции. Он, наряду с лизосомальнойаутофагией , участвует в механизмах таких клеточных адаптации, какатрофия (уменьшение размеров клеток и числа функционирующих структур в них при сохранении жизнеспособности клетки) игипоплазия (уменьшение органа вследствие уменьшения числа клеток в нем при сохранении его жизнеспособности).
Так, например, показано, что инволютивные изменения в коре надпочечников после гипофизэктомии тормозятся актиномицином Д, а, значит, представляют собой активный процесс реализации некой программы саморазборки клеток.
Для обозначения процесса запрограммированной клеточной гибели, морфологически и патохимически отличного от некробиоза, предложен термин «апоптоз». Основатели учения об апоптозе, в частности, Дж. Керр и соавт, считали понятия «запрограммированная клеточная гибель» и «апоптоз» равнозначными. В последнее время имеется тенденция применять первый термин к процессам устранения клеток в раннем онтогенезе, а понятие апоптоз относить только к программируемой гибели зрелых дифференцированных клеток. Так, указывают на наличие аутофагии и отсутствие разрывов ДНК при эмбриональной клеточной гибели, в отличие от апоптоза зрелых клеток.
Вопрос о соотношении некробиоза и апоптоза и о приуроченности этих механизмов к естественной либо насильственной гибели клеток нуждается в обсуждении. Было бы упрощением сказать, что апоптоз – это исключительно процесс естественной гибели клеток, а некробиоз – насильственной. Деление на эти два процесса далеко не абсолютно. Выше, обсуждая паттерны некробиоза, мы уже много раз вынуждены были упоминать об апоптозе, так как между этими процессами много общего. Дело в том, что в ответ на минимальное повреждение или повреждение, не вызывающее быстрого развития глубокой гипоксии и выраженного энергодефицита, клетки могут включать специальную программу самоуничтожения и реагировать апоптозом. В этом случае, например, при действии ионизирующего излучения или вируса СПИДа, смерть клетки насильственна, но механизм ее не некробиотический, а апоптотический. Тельца Каунсильмена, обнаруживаемые при вирусном гепатите в печени, представляют собой результат апоптоза гепатоцитов под воздействием вирус-индуцированного повреждения. Это также насильственная гибель, но механизм ее не связан с быстропрогрессирующей гипоксией и позволяет клетке успеть включить программу саморазборки. Не подлежит сомнению насильственный характер гибели клеток-мишеней под воздействием фактора некроза опухолей. Однако, несмотря на свое категоричное название, данный биорегулятор вызывает в таргетных клетках не только некроз, но и апоптоз. При реализации некробиоза и апоптоза функционируют многие общие механизмы, например, увеличение цитоплазматической концентрации ионизированного кальция и образование свободных активных кислородных радикалов. Более того, при большей силе и интенсивности действия апоптогенный стимул может вызвать некробиоз, очевидно, вследствие того, что прогрессирующий энергодефицит не дает возможности клеткам реализовать энергетически «дорогую» динамику апоптоза.
Таблица 1
Типовые характеристики основных способов гибели клетки.
|
Некробиоз и некроз |
||
|
Морфологическая картина |
Конденсация и фрагментация цитоплазмы, конденсация и рексис ядра, аутофагия митохондрий, мембраны долго остаются стабильными. Нет перифокального воспаления и демаркации. Формируются апоптозные тельца, которые фагоцитируются. |
Кариопикноз или кариолизис, набухание и последующее сморщивание и кальциноз в митохондриях, раннее разрушение мембран, аутолиз клетки, перифокальное воспаление, демаркационный вал |
|
Патохимия |
Нет выраженного энергодефицита, упорядоченные межнуклеосомные разрывы ДНК, синтез БТШ, АРО-1 и других специальных белков, активация эндонуклеазы. Фрагментация цитоплазмы при участии цитоскелета. Тормозится блокаторами кальциевых каналов и актиномицином Д-1. |
Выраженный энергодефицит, гипергидратация клетки, ацидоз, гидролиз, диффузная деградация хроматина. Прекращение белкового синтеза. Парез и агрегация элементов цитоскелета. |
|
Этиология |
Воздействие умеренно сильных повреждений и специальные не повреждающие триггерные сигналы (гормоны, цитокины) |
Воздействие мощных экстремальных факторов |
Если некробиоз всегда сопровождается освобождением в окружающую ткань, а при массивном поражении – и в системный кровоток, медиаторов воспаления, в частности, липидных продуктов деструкции клеточных мембран, то апоптоз протекает без лейкоцитарной демаркации и перифокального воспаления, так как его механизм позволяет избежать значительного выделения медиаторов клеточного повреждения. Издание в 1996 году всеобъемлющей монографии, посвященной апоптозу, облегчает нашу задачу и делает возможным охарактеризовать в данной книге лишь наиболее общие и патофизиологически важные аспекты этой проблемы.
1. Устранение клеток в раннем онтогенезе.
2. Физиологическая инволюция и уравновешивание митозов в зрелых тканях и клеточных популяциях
3. Реализация процессов атрофии и регрессия гиперплазии
4. Альтруистический суицид мутантных и пораженных вирусами клеток
5. Клеточная гибель после слабого воздействия агентов, вызывающих при массированном поражении некроз.
Чтобы более наглядно представить отличия некробиоза и апоптоза, авторы предлагают подробно изучить приводимую таблицу.
Важно отметить, что некроз происходит после насильственной гибели клетки в результате каких-либо причин, вызывающих глубокую тканевую гипоксию, и всегда содержит литический компонент в виде либо лизосомального аутолиза, либо гетеролиза, вызываемого гидролазами фагоцитов. По современным представлениям, аутолиз при гибели клетки носит посмертный характер, а не является элементом некробиоза. Тем не менее, раннее и значительное повреждение клеточных мембран – неотъемлемая часть процессов некробиоза, и практически не наблюдается при апоптозе.
Апоптоз – генетически управляемый процесс, который может быть включен различными пусковыми сигналами без какого-либо существенного предварительного повреждения исполнительного аппарата клетки, хотя может и включиться после умеренного повреждения как альтруистическое самоубийство. Устранение клеток без повреждения возможно и при экспрессии антигена стареющих клеток. Возможно, что эти механизмы «ухода без скандала» комбинируются и/или взаимодействуют.
Принципиально важно, что при неспособности вступить в апоптоз возникает неограниченно пролиферирующий клон клеток, что ведет к серьезным нарушениям в многоклеточном организме и наблюдается, например, при онкологических заболеваниях. До сих пор в данной книге мы часто упоминали об относительной полезности и потенциальной патогенности различных запрограммированных защитных процессов и приводили примеры такой «вредной полезности». В данном случае мы видим основное противоречие патофизиологии, как бы, в обратном ракурсе. Иными словами, апоптоз в клеточном цикле выступает как минимальное запрограммированное зло и также иллюстрирует основное положение наших рассуждений, так как является приспособительной смертью, гибелью по программе и своего рода «полезным вредом» в чистом виде. В любом случае, наблюдения за злокачественными клетками, утратившими под действием онкогенов способность к апоптозу, доказывают, что для клеток утрата способности вовремя умереть – большое зло.
Апоптоз может начаться как ответ генов, программирующих клеточную саморазборку, на рецепторно-опосредоваиный сигнал (например, при стимуляции соответствующими биорегуляторами рецепторов ФНОили глюкокортикоидного рецептора лимфоцитов).
Не только ФНО и глюкокортикоиды, но и почти все цитокины, включая 13 интерлейкинов и 3 интерферона могут быть кодовыми сигналами апоптоза, причем в одних клетках они его запускают, а в других – ингибируют. Тканеспецифические факторы роста и гемопоэтины являются ингибиторами апоптоза для своих клеток-мишеней. Тропные гормоны гипофиза оказывают свой трофический эффект на железы-мишени также путем ингибирования апоптоза.
Сигнал может оказывать на клетку разнонаправленное в отношении апоптоза действие в зависимости от исходного состояния мишени, как это описано выше для ФНО.
В роли генетических индукторов апоптоза, срабатывающих в ответ на рецепторный сигнал, могут выступать гены FAS/АРО-1, с-мус, мах, р53,ced-З и другие. Подавление экспрессии некоторых генов, например,bcl-2, также вызывает апоптоз. Детальное изучение механизмов, с помощью которых продукты этих генов запускают или сдерживают апоптоз, только начато. Однако уже выяснено, что они могут усиливать образование активных кислородных радикалов (как белок АРО-1, гомологичный рецептору фактора некроза опухолей), регулировать перенос кальция в цитоплазму (как продукт генаbcl-2), запускать нейтральные протеазы цитозоля (как продукт генаced-3), связываться с ДНК (как димер белков мус-мах).
Принципиально важно, что апоптоз может быть индуцирован даже в безъядерных постклеточных структурах. Следовательно, первичным звеном апоптоза могут быть не только ядерные события, но и определенные метаболические изменения в цитоплазме или активация долгоживущих матричных РНК, как в случае с антигеном стареющих клеток.
Инициировать апоптоз могут активные кислородные радикалы (АКР). При умеренных повреждениях клетки в отсутствие гипоксии происходит редукция трансмембранного потенциала митохондрий и генерация ими АКР. Если антиоксидантные системы клетки не компенсируют сдвига редокс-потенциала, процесс прогрессирует. При условии отсутствия выраженного энергодефицита и сохранности генетического аппарата реализуется апоптоз, но глубокая гипоксия и выраженные повреждения ДНК инициируют некробиоз. При развитии апоптоза АКР изменяют условия взаимодействия кальция с кальмодулином и способствуют нарастанию цитоплазматической и внутриядерной активности (а при блокаде гена bcl-2 – и росту внутриклеточной концентрации) кальция.
Кальций-зависимое звено механизма апоптоза активирует кальпаины, что ведет к протеолизу белков цитоскелета, образованию цитоплазматических выпячиваний, разрушению межнуклеосомных связей в ядре. Активируется кальцийзависимая эндонуклеаза. Это провоцирует упорядоченные межнуклеосомные разрывы хроматина и фрагментацию ядра. Кальций-зависимая трансглютаминаза агрегирует цитозольные белки. Конечным этапом процесса служит распад клетки на апоптотические тельца и их аутофагоцитоз.
Апоптоз
Смерть клеток в организме может происходит 2 путями: некроза и апоптоза .
Апоптоз – это такой тип гибели клеток, при котором сама клетка активно участвует в процессе своей гибели, т.е. происходит самоуничтожение клетки. Апоптоз, в отличие от некроза, является процессом активным, после воздействия этиологических факторов запускается генетически запрограммированный каскад реакций, сопровождающийся активацией определенных генов, синтезом белков, ферментов, приводящих к эффективному и быстрому удалению клетки из ткани.
Причины апоптоза.
1. Во время эмбриогенеза апоптоз играет важную роль в разрушении различных тканевых зачатков и формировании органов.
2. Апоптозу подвергаются стареющие клетки, закончившие цикл своего развития, например, исчерпавшие запас цитокинов лимфоциты.
3. В растущих тканях определенная часть дочерних клеток подвергается апоптозу. Процент погибающих клеток может регулироваться системными и местными гормонами.
4. Причиной апоптоза может быть слабое воздействие повреждающих факторов, которые при большей интенсивности могут привести к некрозу (гипоксия, ионизирующее излучение, токсины и др.)
Патогенез апоптоза:
Клетка подвергается апоптозу, если в ядре происходит повреждение ДНК, которое не может быть исправлено системой репарации. За данным процессом следит белок, кодируемый геном р53. При невозможности устранения дефекта ДНК под действием р53-протеина активируется программа апоптоза.
На многих клетках имеются рецепторы, воздействие на которые вызывает активацию апоптоза. Наиболее изученными являются Fas-рецептор, обнаруженный на лимфоцитах, и рецептор к фактору некроза опухолей-альфа (TNF-α), обнаруженный на многих клетках. Данные рецепторы играют большую роль в удалении аутореактивных лимфоцитов и регуляции постоянства размера клеточной популяции по типу обратной связи.
Активировать апоптоз могут различные метаболиты и гормоны: противовоспалительные цитокины, стероидные гормоны, окись азота (NO) и свободные радикалы.
Апоптоз клеток активируется при недостатке кислорода в тканях. Причиной его активации может быть действие свободных радикалов, нарушение энергетически зависимых процессов репарации ДНК и др.
Апоптозу подвергаются клетки, утратившие связь с межклеточным матриксом, базальной мембраной или соседними клетками. Утрата данного механизма апоптоза в опухолевых клетках приводит к появлению способности метастазировать.
Некоторые вирусные белки могут активировать апоптоз клеток после самосборки вируса в зараженной клетке. Поглощение апоптотических телец соседними клетками ведет к их заражению вирусом. Вирус СПИДа также может активировать апоптоз незараженных клеток, имеющих на своей поверхности CD4-рецептор.
Также существуют факторы, препятствующие апоптозу. Замедлять апоптоз могут многие метаболиты и гормоны, например, половые гормоны, провоспалительные цитокины. Апоптоз может резко замедляться при дефектах в механизме гибели клетки, например, при мутации в гене р53 или активации генов, тормозящих апоптоз (bcl-2). Многие вирусы обладают способностью ингибировать апоптоз после встраивания собственной ДНК в геном клетки на период синтеза собственных структурных белков.
Морфологические проявления апоптоза
Апоптоз имеет свои отличительные морфологические признаки, как на светооптическом, так и на ультраструктурном уровне. Наиболее четко морфологические признаки выявляются при электронной микроскопии. Для клетки, подвергающейся апоптозу характерно:
Сжатие клетки. Клетка уменьшается в размерах; цитоплазма уплотняется; органеллы, которые выглядят относительно нормальными, располагаются более компактно. Предполагается, что нарушение формы и объема клетки происходит в результате активации в апоптотических клетках трансглютаминазы и цистеиновых протеаз (каспаз). Первая группа ферментов вызывает образование перекрестных связей в цитоплазматических белках, что приводит к формированию своеобразной оболочки под клеточной мембраной, подобно ороговевающим клеткам эпителия, а вторая группа ферментов разрушает белки в цитозоле.
Конденсация хроматина. Это наиболее характерное проявление апоптоза. ДНК расщепляется эндонуклеазами в местах, связывающих отдельные нуклеосомы, что приводит к образованию большого количества фрагментов, в которых число пар оснований делится на 180-200, которые затем конденсируются под ядерной мембраной. Ядро может разрываться на два или несколько фрагментов.
Формирование апоптотических телец. В апоптотической клетке формируются глубокие впячивания клеточной мембраны, что приводит к отшнуровке фрагментов клетки, т.е. формированию окруженных мембраной апоптотических телец, состоящих из цитоплазмы и плотно расположенных органелл, с или без фрагментов ядра.
Фагоцитоз апоптотических клеток или телец осуществляется окружающими здоровыми клетками, как макрофагами, так и паренхиматозными. Апоптотические тельца быстро разрушаются в лизосомах, а окружающие клетки либо мигрируют, либо делятся, чтобы заполнить освободившееся после гибели клетки пространство.
При окраске гематоксилином и эозином апоптоз определяется в единичных клетках или небольших группах клеток. Апоптотические клетки имеют округлую или овальную форму, интенсивно эозинофильную цитоплазму с плотными фрагментами ядерного хроматина. Поскольку сжатие клетки и формирование апоптотических телец происходит быстро и также быстро они фагоцитируются, распадаются или выбрасываются в просвет органа, то на гистологических препаратах апоптоз обнаруживается в случаях его значительной выраженности. К тому же апоптоз – в отличие от некроза – никогда не сопровождается воспалительной реакцией, что также затрудняет его гистологическое выявление.
Для выявления клеток, находящихся на ранних этапах апоптоза, используют специальные иммуногистохимические исследования, например, определение активированных каспаз или TUNEL-метод, визуализирующий разорванную эндонуклеазами ДНК.
Значение апоптоза.
1. Апоптоз имеет огромное значение в эмбриогенезе (включая имплантацию и органогенез). Нарушение гибели клеток в межпальцевых промежутках может привести в синдактилии, а отсутствие апоптоза избыточного эпителия при слиянии небных отростков или тканей, окружающих нервную трубку, приводит к нарушению слияния тканей с двух сторон, что проявляется расщеплением твердого неба и дефектом в тканях, ограничивающих спинномозговой канал, (spina bifida), соответственно.
2. Апоптоз играет важную роль в поддержании постоянства клеточного состава, особенно в гормон-чувствительных тканях. Замедление апоптоза приводит к гиперплазии тканей, ускорение – к атрофии. Он принимает участие в отторжении эндометрия во время менструального цикла, атрезии фолликулов в яичниках в менопаузе и регрессии ткани молочной железы после прекращения лактации.
3. В данный момент исследуется огромное количество лекарственных препаратов, направленных на регуляцию апоптоза в определенных тканях. Так, ускорение апоптоза иммунокомпетентных клеток можно использовать для лечения аутоиммунных заболеваний и предотвращения отторжения трансплантата, замедление апоптоза может использоваться для предотвращения апоптоза в тканях, испытывающих ишемию, повышенное внешнее давление или временно бездействующих тканях. Замедление апоптоза при вирусных инфекциях предотвращает распространение инфекции на соседние клетки.
4. Во всех опухолях наблюдается нарушение апоптоза в опухолевых клетках. Эта поломка может происходить на разных этапах апоптоза, например, может происходить мутация гена р53, что приведен к тому, что мутантный протеин-р53 будет накапливаться в клетке в избыточном количестве, но не будет вызывать апоптоз несмотря на дефекты в геноме клетки, что приведет к пролиферации клеток с нарушенным геномом, причем с каждым последующим делением нарушения в ДНК будут накапливаться. Иногда в опухолевых клетках может накапливаться и нормальный, или «дикий», протеин р53, если поломка в механизме апоптоза происходит на других уровнях. При хронических лимфоидных лейкемиях наблюдается накопление продуктов гена bcl-2, что приводит к патологическому удлинению срока жизни опухолевых клеток и резистентности клеток к различным проапоптотическим факторам. Иногда нарушается передача сигналов от рецепторов гибели клеток, например, с рецептора к TNF-α. TNF-α принимает участие в регуляции клеточной популяции по типу обратной связи. Все клетки в популяции выделяют TNF-α в небольших количествах; чем больше клеток в ткани, тем выше концентрация TNF-α, а значит и уровень апоптоза. Таким образом достигается баланс между пролиферацией и гибелью клеток. Опухолевые клетки теряют способность подвергаться апоптозу под действием этого цитокина, при этом он накапливается в большом количестве в опухолевой ткани. В результате этого TNF-α начинает в большом количестве попадать в кровоток и вызывать апоптоз паренхиматозных клеток во многих органах, приводя к кахексии.
Процесс, при котором клетка может убивать сама себя, называется запрограммированной клеточной гибелью (ЗГК). Этот механизм имеет несколько разновидностей и играет важнейшую роль в физиологии различных организмов, особенно многоклеточных. Самой часто встречающейся и хорошо изученной формой ЗГК является апоптоз.
Что такое апоптоз
Апоптоз - это контролируемый физиологический процесс самоуничтожения клетки, характеризующийся поэтапным разрушением и фрагментацией ее содержимого с формированием мембранных пузырьков (апоптозных телец), впоследствии поглощаемых фагоцитами. Этот генетически заложенный механизм активируется под воздействием определенных внутренних или внешних факторов.
При таком варианте гибели клеточное содержимое не выходит за пределы мембраны и не вызывает воспаление. Нарушения в регуляции апоптоза приводят к серьезным патологиям, таким как неконтролируемые клеточные деления или дегенерация тканей.
Апоптоз представляет собой лишь одну из нескольких форм запрограммированной гибели клетки (ЗГК), поэтому отождествлять эти понятия ошибочно. К известным видам клеточного самоуничтожения относят также митотическую катастрофу, аутофагию и программированный некроз. Другие механизмы ЗГК пока не изучены.
Причины апоптоза клеток
Причиной запуска механизма запрограммированной клеточной гибели могут быть как естественные физиологические процессы, так и патологические изменения, вызванные внутренними дефектами или воздействием внешних неблагоприятных факторов.
В норме апоптоз уравновешивает процесс деления клеток, регулируя их количество и способствуя обновлению тканей. В таком случае причиной ЗГК служат определенные сигналы, входящие в систему контроля гомеостаза. С помощью апоптоза уничтожаются одноразовые или выполнившие свою функцию клетки. Так, повышенное содержание лейкоцитов, нейтрофилов и других элементов клеточного иммунитета по окончании борьбы с инфекцией устраняется именно за счет апоптоза.
Запрограммированная гибель является частью физиологического цикла репродуктивных систем. Апоптоз задействован в процессе оогенеза, а также способствует гибели яйцеклетки при отсутствии оплодотворения.
Классическим примером участия апоптоза клеток в жизненном цикле вегетативных систем является осенний листопад. Сам термин происходит от греческого слова apoptosis, что буквально переводится как "опадание".
Апоптоз играет важнейшую роль в эмбриогенезе и онтогенезе, когда в организме сменяются ткани и атрофируются определенные органы. Примером могут служить исчезновение перепонок между пальцами конечностей некоторых млекопитающих или отмирание хвоста при метаморфозе лягушки.
Апоптоз может быть спровоцирован накоплением дефектных изменений в клетке, возникших в результате мутаций, старения или ошибок митоза. Причиной запуска ЗГК могут быть неблагоприятная среда (недостаток питательных компонентов, дефицит кислорода) и патологические внешние воздействия, опосредованные вирусами, бактериями, токсинами и т. д. При этом если повреждающий эффект слишком интенсивен, то клетка не успевает осуществить механизм апоптоза и погибает в результате развития патологического процесса - некроза.

Морфологические и структурно-биохимические изменения клетки во время апоптоза
Процесс апоптоза характеризуется определенным набором морфологических изменений, которые с помощью микроскопии можно наблюдать в препарате ткани in vitro.

К основным признакам, характерным для апоптоза клеток, относят:
- перестраивание цитоскелета;
- уплотнение клеточного содержимого;
- конденсацию хроматина;
- фрагментацию ядра;
- уменьшение объема клетки;
- сморщивание контура мембраны;
- образование пузырьков на клеточной поверхности,
- деструкцию органоидов.
У животных эти процессы завершаются образованием апоптоцитов, которые могут быть поглощены как макрофагами, так и соседними клетками ткани. У растений формирования апоптозных телец не происходит, а после деградации протопласта сохраняется остов в виде клеточной стенки.

Помимо морфологических изменений, апоптоз сопровождается рядом перестроек на молекулярном уровне. Происходит повышение липазной и нуклеазной активностей, которые влекут за собой фрагментацию хроматина и многих белков. Резко увеличивается содержание сАМФ, изменяется структура клеточной мембраны. В растительных клетках наблюдается образование гигантских вакуолей.
Чем апоптоз отличается от некроза

Главное различие между апоптозом и некрозом заключается в причине клеточной деградации. В первом случае источником разрушения служат молекулярные инструменты самой клетки, которые работают под строгим контролем и требуют затрат энергии АТФ. При некрозе происходит пассивное прекращение жизнедеятельности из-за внешнего повреждающего воздействия.
Апоптоз - это естественный физиологический процесс, сконструированный таким образом, чтобы не вредить окружающим клеткам. Некроз - это неконтролируемое патологическое явление, возникающее в результате критических повреждений. Поэтому неудивительно, что механизм, морфология и последствия апоптоза и некроза во многом противоположны. Однако имеются и общие черты.
В случае повреждения клетки запускают механизм запрограммированной гибели в том числе для того, чтобы не допустить некротического развития. Однако недавние исследования показали, что существует иная непатологическая форма некроза, которую также отнесли к ЗГК.
Биологическое значение апоптоза
Несмотря на то что апоптоз приводит к клеточной гибели, его роль для поддержания нормальной жизнедеятельности всего организма очень велика. Благодаря механизму ЗГК осуществляются следующие физиологические функции:
- поддержание баланса между пролиферацией и смертью клеток;
- обновление тканей и органов;
- устранение дефектных и "старых" клеток;
- защита от развития патогенного некроза;
- смена тканей и органов при эмбрио- и онтогенезе;
- удаление ненужных элементов, выполнивших свою функцию;
- устранение клеток, нежелательных или опасных для организма (мутантных, опухолевых, зараженных вирусом);
- предотвращение развития инфекции.
Таким образом, апоптоз является одним из способов поддержания клеточно-тканевого гомеостаза.

Этапы клеточной смерти
То, что происходит с клеткой при апоптозе, является результатом сложной цепочки молекулярных взаимодействий между различными ферментами. Реакции проходят по типу каскада, когда одни белки активируют другие, способствуя постепенному развитию сценария гибели. Этот процесс можно разделить на несколько этапов:
- Индукция.
- Активация проапоптических белков.
- Активация каспаз.
- Разрушение и перестройка клеточных органелл.
- Формирование апоптоцитов.
- Подготовка клеточных фрагментов к фагоцитозу.
Синтез всех компонентов, необходимых для запуска, реализации и контроля каждого этапа заложен генетически, почему апоптоз и называют запрограммированной гибелью клетки. Активация этого процесса находится под строгим контролем регуляторных систем, включающих в том числе и различные ингибиторы ЗГК.
Молекулярные механизмы апоптоза клетки
Развитие апоптоза обуславливается совокупным действием двух молекулярных систем: индукционной и эффекторной. Первый блок отвечает за контролируемый запуск ЗГК. В него входят так называемые рецепторы смерти, Cys-Asp-протеазы (каспазы), ряд митохондриальных компонентов и проапоптических белков. Все элементы индукционной фазы можно разделить на тригеры (участвуют в индукции) и модуляторы, обеспечивающие трансдукцию сигнала смерти.
Эффекторную систему составляют молекулярные инструменты, обеспечивающие деградацию и перестройку клеточных компонентов. Переход между первой и второй фазами осуществляется на этапе протеолитического каспазного каскада. Именно за счет компонентов эффекторного блока происходит гибель клетки при апоптозе.
Факторы апоптоза
Структурно-морфологические и биохимические изменения при апоптозе осуществляются определенным набором специализированных клеточных инструментов, среди которых наиболее важными являются каспасы, нуклеазы и мембранные модификаторы.
Каспазы - группа ферментов, разрезающих пептидные связи по остаткам аспарагина, фрагментируя белки на крупные пептиды. До начала апоптоза присутствуют в клетке в неактивном состоянии из-за ингибиторов. Главной мишенью каспаз являются ядерные белки.
Нуклеазы - ответственны за разрезание молекул ДНК. Особо важна в развитии апоптоза активная эндонуклеаза CAD, разрывающая участки хроматина в областях линкерных последовательностей. В результате образуются фрагменты длиной 120-180 нуклеотидных пар. Комплексное воздействие протеолитических каспаз и нуклеаз приводит к деформации и фрагментации ядра.

Модификаторы клеточной мембраны - нарушают асимметричность билипидного слоя, превращая его в мишень для фагоцитирующих клеток.
Ключевая роль в развитии апоптоза принадлежит каспазам, которые поэтапно активируют все последующие механизмы деградации и морфологической перестройки.
Роль каспаз в клеточной гибели
Семейство каспаз включает 14 белков. Часть из них не задействована в апоптозе, а остальные подразделяются на 2 группы: инициаторные (2, 8, 9, 10, 12) и эффекторные (3, 6 и 7), которые иначе называются каспазами второго эшелона. Все эти белки синтезируются в виде предшественников - прокаспаз, активируемых протеолитическим расщеплением, суть которого состоит в отсоединении N-концевого домена и разделении оставшейся молекулы на две части, в последствии ассоциирующиеся в димеры и тетрамеры.
Инициаторные каспазы необходимы для активации эффекторной группы, которая проявляет протеолитическую активность в отношении различных жизненно важных клеточных белков. К субстратам каспаз второго эшелона относятся:
- ферменты репарации ДНК;
- игибитор белка р-53;
- поли-(ADP-рибозо)-полимераза;
- ингибитор ДНК-азы DFF (разрушение этого белка приводит к активации эндонуклеазы CAD) и др.
Общее количество мишеней эффекторных каспаз насчитывает более 60 белков.
Ингибирование апоптоза клеток еще возможно на стадии активации инициаторных прокаспаз. Когда эффекторные каспазы вступают в действие, процесс становится необратимым.
Пути активации апоптоза
Передача сигнала для запуска апоптоза клетки может быть осуществлена двумя путями: рецепторным (или внешним) и митохондриальным. В первом случае процесс активируется через специфические рецепторы смерти, воспринимающие внешние сигналы, которыми служат белки семейства TNF или Fas-лиганды, расположенные на поверхности Т-киллеров.
В состав рецептора входит 2 функциональных домена: трансмембранный (предназначенный для связи с лигандом) и ориентированный внутрь клетки "домен смерти", индуцирующий апоптоз. Механизм рецепторного пути основывается на образовании DISC-комплекса, активирующего инициаторные каспазы 8 или 10.
Сборка начинается со взаимодействия домена смерти с внутриклеточными адапторными белками, которые, в свою очередь, связывают инициаторные прокаспазы. В составе комплекса последние превращаются в функционально-активные каспазы и запускают дальнейший апоптозный каскад.
Механизм внутреннего пути основан на активации протеолитического каскада особыми митохондриальными белками, выброс которых контролируется внутриклеточными сигналами. Выход компонентов органоидов осуществляется через образование огромных пор.
Особая роль в запуске принадлежит цитохрому с. Попадая в цитоплазму, этот компонент электротранспортной цепи связывается с белком Apaf1 (апоптотический фактор активации протеаз), что приводит к активации последнего. Затем Apaf1 связывают инициаторные прокаспазы 9, которые по механизму каскада запускают апоптоз.
Контроль внутреннего пути осуществляется особой группой белков семейства Bcl12, которые регулируют выход межмембранных компонентов митохондрий в цитоплазму. В составе семейства имеются как проапоптические, так и антиапоптические белки, баланс между которыми и определяет, будет ли запущен процесс.
К одним из мощных факторов, запускающих апоптоз по митохондриальному механизму, относятся реактивные формы кислорода. Еще одним значимым индуктором является белок р53, который активирует митохондриальный путь при наличии ДНК-повреждений.
Иногда запуск апоптоза клеток сочетает в себе сразу два пути: как внешний, так и внутренний. Последний обычно служит для усиления рецепторной активации.