Ювенильная миопатия эрба. Прогрессирующая мышечная дистрофия Эрба—Рота
Читайте также
В современной неврологии существует огромное количество заболеваний, характер возникновений которых не поддаются рациональному объяснению со стороны специалистов. К таким болезням можно отнести группу таких недугов, как мышечная дистрофия. Всего разновидностей данной болезни девять, но обо всем по порядку…
Мышечная дистрофия - это хроническое наследственное заболевание в результате развития которого происходит поражение мышечной системы человека. Пораженные мышцы перестают нормально функционировать, истончаются в размерах, а в месте их расположения в организме постепенно нарастает жировая прослойка.
Разновидности мышечной дистрофии
В современной неврологии данный недуг классифицируется на девять различных болезней. Классификация заболевания связана с:
- локализацией мышечных нарушений;
- характеристиками болезни;
- агрессивностью развития;
- возрастом.
Так, мышечная дистрофия бывает:
- Дюшенна;
- миотоническая (болезнь Штейнерта);
- Беккера;
- Эрба Рота;
- юношеская форма дистрофии Эрба-Рота;
- плече - лопаточная лицевая форма (Ландузи-Дежерина);
- алкогольная миопатия;
- дистальная форма;
- миодистрофия Эмери -Дрейфуса.
Дистрофия Дюшенна
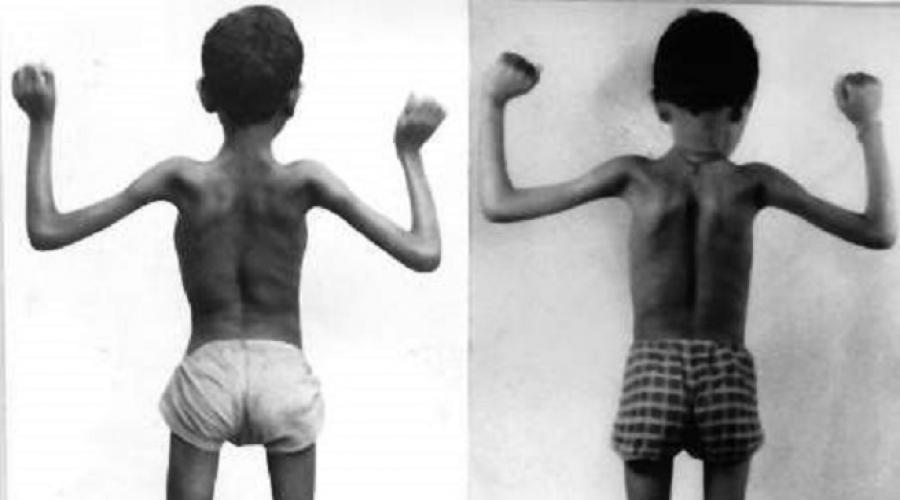
Наиболее известная форма прогрессирующая дистрофия Дюшенна (псевдогипертрофическая дистрофия, мерозин - негативная врожденная дистрофия). Данная разновидность недуга проявляется в детском возрасте начиная с двух до пяти лет. В первую очередь от данного недуга страдают мышцы нижних конечностей, которые несмотря на малоподвижный образ жизни у маленьких пациентов, постепенно увеличиваются в размерах. Данная особенность связана с нарастанием жировой ткани вместо мышц.

Слева здоровый ребенок, справа больной
Постепенно, по мере прогрессирования болезни она переходит в верхнюю часть и поражает мышцы верхних конечностей. Как правило, уже к двенадцатилетнему возрасту маленький пациент перестает полностью двигаться. Летальность у данного заболевания очень высока примерно 85–90% больных не доживают до 20-летнего возраста.
В группе риска находятся представители мужского пола, так как девочек данный недуг практически не затрагивает.
Болезнь Штейнерта
Врожденная дистрофия Штейнера не зря имеет название миотоническая, так как в результате прогрессирования болезни происходит слишком медленное расслабление мышц после их сокращения (такое явление называется миотонией).
Данное заболевание, в отличие от предыдущего распространено у взрослых, в возрасте от 20 до 40 лет. Имеют место быть и случаи прогрессирования болезни у детей, как правило, в младенческом возрасте, однако, это скорее исключение, чем правило.

На лицо признаки миотонии (приоткрытый рот и веки)
Гендерной зависимости болезнь не имеет и в равной степени от нее страдают как мужчины, так и женщины. Отмечается, что при недуге проявляется слабость мимических мышц лица, а также конечностей. Прогрессирование, в отличие от болезни Дюшенна происходит медленно.
Отличительная черта заболевания, возможность поражения не только мышц конечностей, но и внутренних мышц (сердечная мышца), что в свою очередь, представляет большую опасностью для жизни человека.
Болезнь Беккера
Данная форма болезни также долго прогрессирует, и пациент длительное время чувствует себя хорошо. Обострение недуга может произойти на фоне полученных травм или различных заболеваний нервной системы, которые своим течением ускорят развитие недуга.
В группе риска находятся люди низкого роста.
Болезнь Эрба-Рота и юношеская ее форма
Данное аутосомно - рецессивное заболевание развивается у пациентов после 20 лет, а юношеская форма у детей и подростков 11-13-летнего возраста. Прогрессирование болезни происходит по восходящему варианту, то есть сначала поражаются мышцы нижних конечностей, и постепенно происходит восхождение недуга к верхним конечностям.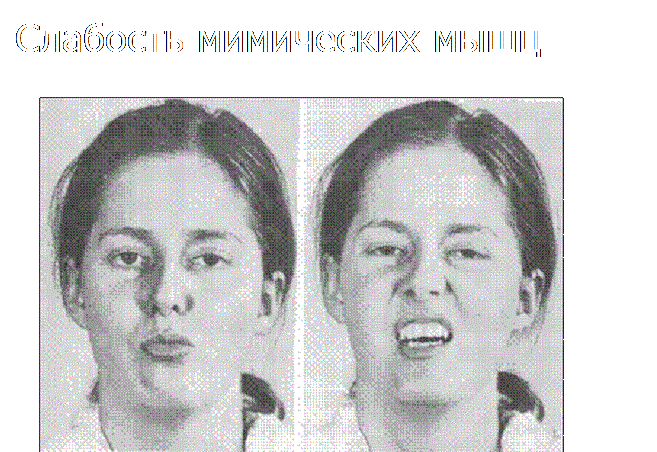
Отличительной чертой недуга является наличие выступающих лопаток, которые по мере прогрессирования выступают более явно и очевидно.
Во время ходьбы отмечается переваливание пациента, выпячивание живота и задвигание грудной клетки.
Болезнь Ландузи-Дежерина
Плече-лопаточно-лицевая форма данного заболевания является наиболее безразборным видом, так как поражает людей в возрасте от пяти до 55 лет. Для данного недуга характерно очень длительное развитие, пациент может сохранять трудоспособность до 25 лет жизни с данной болезнью.
Отличительными симптомами является поражение лицевых мышц, в результате чего у больного могут возникнуть проблемы с четкостью произношения, в связи с неполным смыканием губ. Кроме того, отмечается неполное смыкание век.
По мере развития у больного атрофируются лицевые мышцы, мышцы плеч, конечности и туловища.
Данная форма не зависит от генетической мутации.
Алкогольная миопатия
Данный вид болезни также несвязан с генными мутациями и причина его возникновения одна - злоупотребление алкоголем. У больных могут отмечаться болевые синдромы в конечностях, связанные именно с поражением мышц.
Различают острую и хроническую алкогольную миопатию.
Дистальная форма
Дистальная форма мышечной дистрофии является доброкачественным вариантом прогрессирующей дистрофии. Как правило, данное заболевание трудно дифференцировать от невральной амиатрофии Мари-Шарко. Для разделения этих двух заболеваний требуется проведение электроэнцефалограмму головы, которая и дает понимание с какой болезнью предстоит иметь дело.
К основным симптомам недуга относят атрофию мышц конечностей с их последующим исхуданием. Возможны парезы стоп, кистей рук и т. п.
Миодистрофия Эмери -Дрейфуса
Данный тип болезни первоначально, вообще, не был выделен в качестве отдельного заболевания, так как по своей симптоматике был схож с дистрофией Дюшенна. Однако, в дальнейшем в результате длительного исследования было установлено, что болезнь Эмери -Дрейфуса обладает индивидуальной симптоматикой.
Болезнь относят в разряд редких. В группе риска находятся люди до 30 лет, как правило, юный возраст. Есть свидетельства о проявлении симптомов и после 30 лет, но они единичны.
Основное отличие данного вида болезни - это проблемы с мышцами сердца, которые в итоге могут стать причиной смерти. Кардиомиопатия при данном недуге не единственное отличие. Кроме проблем с сердцем, у пациентов наблюдают стандартные для дистрофии Дюшенна признаки, но в более щадящем режиме развития.
Причины
Негативная составляющая болезней нервной системы заключается в том, что они тяжело поддаются изучению. По этой причине не до конца известно о факторах, провоцирующих развитие той или иной формы мышечной дистрофии.
Основная причина, для формирования большинства подвидов данной болезни является генная мутация, в частности гена, который отвечает за синтез белка.
К примеру, болезнь Дюшенна имеет прямое отношение к мутации половой Х хромосомы. Основной переносчик нехорошего гена выступают девушки, которые несмотря на его наличие в собственной ДНК не страдают от данной болезни.
Что касается миотонической формы, то виновник ее возникновения ген, расположенный в 19 хромосоме.
Основные симптомы
Наличие большого количество различных подвидов у данного недуга указывает на различия в симптоматике, однако у болезни есть общие симптомы, которые в себя включают:

Диагностика мышечной дистрофии
Диагностические мероприятия при подобном недуге обширны, так как существует большое количество болезней, от которых необходимо дифференцировать болезнь.
На первоначальном этапе доктор обязательно изучит анамнез больного, уточнит о симптоматике, режиме дня и т. п. Эти данные требуются для составления плана проведения последующих диагностических мер, которые могут в себя включать:

Стоит отметить, что чем позже проявилась болезнь, тем лучше для больного, так как ранние симптомы в большинстве случаев заканчиваются смертью.
Лечение
Лечение мышечной дистрофии процесс сложный и затяжной, однако, в настоящее время не создано лекарство, которое полностью исцеляет больного. Все мероприятия направлены на облегчение жизни больного и восстановления некоторых утраченных способностей.
Для затормаживания развития болезни назначают следующие препараты:
- кортикостероиды;
- витамины В1;
- аденозинтрифосфат (АТФ).
Кроме того, для замедления процесса развития используют фетальные стволовые клетки, которые замедляют процесс дистрофии.
Помимо того, в качестве профилактических мероприятий назначают:
- массаж;
- физиотерапию;
- дыхательную гимнастику.
Помимо стандартных вариантов лечения, важно постоянно руководствоваться тремя главными составляющими в процессе жизнедеятельности:
- Адекватная физическая активность.
- Своевременная психологическая поддержка.
- Соблюдение диеты.
Физическая активность
Отсутствие желания у человека бороться с недугом оказывает негативное воздействие на организм. Судите сами, пассивность, нежелание двигаться угнетает и без того пораженную мышечную систему. Мышцам необходимо давать нагрузку, так как без нагрузки дистрофичные процессы начинают происходить быстрее, тем самым прогрессируя в более быстром темпе.
Умеренная физическая активность, использование поддерживающих приспособлений будут отличным подспорьем в борьбе с недугом.
При наличии болей в мышцах отлично подойдет плавание, йога, упражнения на растяжку.
Психологическая поддержка
Психологическая поддержка окружения важна для больного человека. А если недуг такой серьезный, как этот тем более. Для кого-то будет достаточно обычной поддержки со стороны друзей и родных, а кому-то может потребоваться квалифицированная психологическая помощь.
Важно дать понять такому человеку, что он не остался один на один со своей проблемой. Он должен понимать, что ему есть к кому обратиться, есть люди, которые сопереживают и поддерживают его.
Диета
Что касается режима питания и соблюдения диеты, существует расхожее мнение, что соблюдение противовоспалительной диеты может замедлить прогрессирование недуга. Данная диета снижает воспаление, уровень глюкозы, выводит токсины из организма и питает его полезными веществами.
Суть такой диеты в следующем:
- Отказ от продуктов, содержащих «плохие» жиры и замена их на «хорошие», введение в рацион ненасыщенных жиров, которые содержатся в оливковом, льняном, кунжутном масле, авокадо.
- Применение в пищу мяса и рыбы, при производстве которых не были использованы антибиотики или гормоны.
- Полное удаление из рациона рафинированного сахара и глютена.
- Употребление в пищу следующих продуктов - китайская капуста, брокколи, сельдерей, ананас, лосось, свекла, кукумария, имбирь, куркума и других продуктов, обладающих противовоспалительными свойствами.
- Молочные продукты допустимы только на основе овечьего и козьего молока.
- Допустимо употребление травяных чаев, лимонада, кваса, морсов и натуральных соков.
Профилактика
Так как мышечную дистрофию довольно тяжело предсказать и обнаружить на раннем этапе, профилактические мероприятия сводятся к двум простым рекомендациям:
Обязательное обследование женщины на этапе планирования беременности на предмет наличия мутаций в генах
В случае если до беременности, по каким-либо причинам не было сделано генетического обследования, уже в период беременности проводится тест на определение у плода мутаций в Х хромосоме.
Прогноз и осложнения
В зависимости от вида болезни прогноз может быть разным, и тем не менее можно выделить несколько возможных осложнений, возникающих при данном недуге.
- нарушения сердечной деятельности
- искривление позвоночника
- снижение интеллектуальных способностей больного
- снижение двигательной активности
- нарушения дыхательной системы
- летальный итог
Итак, мышечная дистрофия довольно опасное и неизлечимое заболевание, поэтому будущим родителям необходимо с глубокой ответственностью относиться к планированию беременности. Берегите себя и своих будущих детей!
Настаивайте на точной постановке диагноза, изучайте информацию, а статья Клавдии поможет вам в этом.
Мой диагноз — миопатия Эрба-Рота! Точно ли?
Зачем мне точный диагноз? Всё равно это неизлечимо…
Зачем мне дополнительная диагностика? Мой диагноз — миопатия Эрба-Рота!
Мне уже сделали кучу анализов, и там всё было отрицательно…
Знакомы ли вам такие мысли?
Как мы не раз писали, миопатия Эрба-Рота (или ПКМД) — это описательный диагноз. Под этим именем скрываются десятки заболеваний с разными особенностями, прогнозом и потенциалом лечения.
И среди них есть один диагноз, на который нужно обратить особое внимание. Это пока что единственная поясно-конечностная мышечная дистрофия, для которой лечение есть. Эта ПКМД — болезнь Помпе с поздним началом.
Бессмысленно пускаться в описание типичных проявлений болезни Помпе: так много лиц у этого заболевания. У кого-то активность КФК (креатинкиназы) на верхней границе нормы (около 300 ед/л), а у кого-то — под 3000 ед/л. У одного пациента болезнь начинается с одышки и ночных апноэ, а у другого больше страдают скелетные мышцы, но с дыханием всё нормально. Болезнь Помпе с поздним началом может дать о себе знать и в 5 лет, и в 55.
Если вам поставлен диагноз «поясно-конечностная мышечная дистрофия» или «миопатия Эрба-Рота», то у вас может быть болезнь Помпе с поздним началом.
Пройти анализ на болезнь Помпе можно бесплатно . Для этого вам нужно найти врача любой специальности, который согласится вам помочь. Этому врачу нужно позвонить на телефон горячей линии поддержки диагностики болезни Помпе — 88001002494 (о программе диагностики на сайте МГНЦ).
Дополнительная информация : .Анализ делается по сухим пятнам крови. Специалист горячей линии расскажет, как это сделать и где взять необходимые материалы.
Сначала будет выполнен скрининговый ферментный тест: по его результатам будет понятно, исключена ли у вас болезнь Помпе. Если результат покажет, что она не исключена, то лаборатория проведёт ДНК-анализ, а уж его результат поставит точку в вопросе о наличии болезни Помпе.
В Москве это можно сделать, например, в Медико-генетическом научном центре на ул. Москворечье, 1. Если вы живете не в Москве и хотите исключить у себя болезнь Помпе, то напишите Инне Становой — самой активной российской пациентке с болезнью Помпе, она подскажет, как действовать.
Препарат для лечения болезни Помпе — Майозайм — зарегистрирован в России, его бесплатно получают практически все российские пациенты с болезнью Помпе
. Получить его непросто, но оно того стоит, ведь Майозайм — это патогенетическая терапия, то есть та, что действует на причину болезни Помпе, сильно замедляя или останавливая её развитие, а не просто облегчая симптомы.
Не упускайте время! Вдруг у вас именно болезнь Помпе?
Миодистрофия - прогрессирующая атрофия мышц. Наблюдается довольно часто. Заболевают лица обоего пола. Болезнь наследственная. Различают несколько форм миодистрофии, которые наследуются по разным типам и поэтому рассматриваются как самостоятельные заболевания. Из них самым распространенным является юношеская миодистрофия Эрба-Рота, плече-лопаточно-лицевая миодистрофия Ландузи-Дежерина и псевдогипертрофическая миодистрофия Дюшенна.
Юношеская миодистрофия Эрба - Рота
Юношеская миодистрофия Эрба - Рота наследуется по аутосомно-рецессивным типом (болеют дети здоровых родителей). Первые признаки болезни проявляются преимущественно в 14-16 лет, очень редко - в 5-10-летнем возрасте. Начальными симптомами являются мышечная слабость, патологическая мышечная утомляемость при физической нагрузке; походка становится «утиной». Атрофии сначала локализуются в проксимальных группах мышц тазового пояса и нижних конечностей. Иногда миодистрофичний процесс одновременно поражает мышцы тазового и плечевого поясов. В поздних стадиях в процесс вовлекаются мышцы спины и живота. Вследствие атрофии формируются лордоз, крыловидные лопатки, «осиная талия». При вставании больные применяют вспомогательные приемы, опираются руками на бедра. Проявление псевдогипертрофии мышц, контрактуры суставов, сухожильной ретракции незначителен. Сухожильные рефлексы с верхних и нижних конечностей снижены. Течение болезни длительное, медленно прогрессирующее. В возрасте 35-40 лет больные теряют способность самостоятельно передвигаться, рано наступает инвалидность.Плечо-лопаточно-лицевая миодистрофия Ландузи - Дежерина
Плечо-лопаточно-лицевая миодистрофия Ландузи - Дежерина наследуется по аутосомно-доминантному типу.Клинические проявления плечо-лопаточно-лицевой миодистрофии Ландузи - Дежсрина
Первые признаки заболевания проявляются преимущественно в возрасте 10-20 лет. Мышечная слабость и атрофии локализуются преимущественно в области лица, лопаток, плеч. Вследствие атрофии лица становится гипомимичним. Для больных типичны гладкие лоб, лагофтальм, «поперечная» улыбка, толстые, иногда вывернутые губы (губы тапира). Атрофии мышц плеча, большой грудной, переднего зубчатого, трапециевидной мышц обусловливают возникновение симптомов свободных надплич, крыловидных лопаток, увеличение межлопаточной промежутке, уплощение грудной клетки, . Иногда атрофии распространяются на мышцы нижних конечностей. Псевдогипертрофии выраженные в икроножных и дельтовидных мышцах. В ранних стадиях болезни тонус снижен в проксимальных группах мышц. Сухожильные рефлексы снижены (преимущественно с двуглавой и трехглавой мышц плеча). Течение заболевания медленно прогрессирующее. Больные длительное время сохраняют работоспособность.Псевдогипертрофическая миодистрофия Дюшенна
Псевдогипертрофическая миодистрофия Дюшенна - злокачественная из всех болезней нервно-мышечной системы. Это заболевание наблюдается только у мальчиков, так наследуется по рецессивному, сцепленным с Х-хромосомой типу. Передается болезнь по материнской линии, начинается в первые годы жизни. Атрофия мышц вначале локализуется в мышцах тазового пояса и бедер, вследствие чего рано возникают затруднения при ходьбе вверх по лестнице; походка становится «утиной». Дети часто падают, с трудом поднимаются. Изменяется статика. В возрасте до 10-12 лет больные теряют способность самостоятельно передвигаться, они прикованы к постели. При этой форме миодистрофии нарушается интеллект. Происходят изменения и в мышцах сердца. Развиваются псевдогипертрофии икроножных мышц. В основе всех прогрессирующих миодистрофий лежит постепенное перерождение мышечных волокон скелетных мышц и замена их соединительной и жировой тканью. Вследствие этого развиваются ложные гипертрофии мышц, чаще икроножных (при миодистрофия Дюшенна и Эрба) и ретракция (сокращение) ахилловых сухожилий. Дегенерация мышц происходит вследствие патологически измененного обмена веществ в них. Грубо нарушается белковый и углеводный обмен.Лечение псевдогипертрофической миодистрофии Дюшенна
Рекомендуют диету, богатую белками (мясо, рыба, сыр), витамины. Ограничивать движения в верхних конечностях не нужно, наоборот, показаны лечебная гимнастика и массаж. Курсами вводят ретаболил (по 1 мл в неделю, всего 4 инъекции). Показаны АТФ по 1 мл внутримышечно ежедневно (на курс лечения 15-20 инъекций), церебролизин (по 1 мл внутримышечно, 20-ЗО инъекций), а также анаприлин - по 20-40 мг 2 раза в день (в течение 4 нед с постепенной отменой препарата). Рекомендуют также прием глютаминовой кислоты, рибоксина, метионина. Назначают токоферол, ретинол, аскорбиновую кислоту, витамины группы В. Показаны средства, улучшающие микроциркуляцию: никотиновая кислота, ксантинола никотинат, сермюн, актовегин, пентоксифиллин, пармидин. Для улучшения нервно-мышечной проводимости применяют антихолинэстеразные препараты: прозерин, галантамин, оксазил, пиридостигмина бромид, стефаглабрину сульфат. Одновременно назначают лечебную физкультуру, массаж, физиотерапевтические процедуры, иглоукалывание. Важной является профилактика костно-суставных деформаций и контрактур конечностей. Используют тепловые процедуры: озокерит, грязевые аппликации, радоновые, хвойные, сульфидные, сероводородные ванны, оксигенобаротерапию. Показано ортопедическое лечение приПрогрессирующая мышечная дистрофия Эрба-Рота является одной из наследственных форм поражения мышечной ткани.
Начало заболевания обычно в возрасте 10-20 лет (возможен дебют до 40 лет); проходит около 10-15 лет до полного обездвиживания. Передача признака – по рецессивному типу, сцепленному с полом.
Информация для врачей. Мышечная дистрофия Эрба-Рота шифруется по МКБ 10 под кодом G71.0. При этом в диагнозе обязательно указывается стадия заболевания (1 – умеренные нарушения движений, 2 – затруднения возникают при ходьбе, выполнении легкой физической работе, 3 – параличи, контрактуры и т.д.). После этого указывается выраженность сопутствующих проявлений (снижение интеллекта, осложнений со стороны сердца), темп прогрессирования (быстрое, среднее или медленное). При утрате способности к передвижению обязательно указывается этот факт.
Причины
Единой точки рения на возникновение мышечных атрофий не существует. Доминирует наследственная теория. Точные биохимические механизмы патогенеза до конца не раскрыты.
Симптомы
Симптомы заболевания вначале не специфичны и включают в себя общую слабость, слабость мышц спины. Постепенно заболевание прогрессирует. Пациент перестает удерживать спину в нормальном положении, развивается гиперлордоз – переразгибание поясницы кзади.
Достаточно рано изменяется походка. Она становится похожей на «утиную» - переваливание ног из-за слабости мышц бедра и тазового пояса. Быстро развивается гипотрофия мышц верхнего плечевого пояса. Также постепенно развивается общая гипотрофия, а затем и атрофия мышц. Иногда имеет псевдогипертрофия голеней – замена мышечной массы жировой и соединительной тканью.
Со временем пациент перестает выполнять многие активные движения. Значимо затрудняется вставание, больным приходится вставать на четвереньки, помогать руками при вставании. Лицо пациента становится амимично, неполностью смыкаются веки, губы же, напротив, выворачиваются кпереди и нередко утолщаются (губы тапира). Мимику такого пациента ещё иногда называют лицом миопата.
Диагностика
Диагноз заболевания обычно выставляется достаточно точно. При постановке диагноза дистрофия Эрба-Рота обращают внимание на возраст в дебюте заболевания, наследственность, скорость прогрессии процесса. В неврологическом осмотре выявляется снижение рефлексов вплоть до выпадения, снижение тонуса мышц, наличие контрактур суставов (из-за неравномерного процесса атрофии мышц).
Вопреки заблуждениям, фасцикулярных подергиваний мышц не бывает. При регистрации биотоков мышц снижается амплитуда, но не частота разрядов. По ЭНМГ определяется укорочение длительности потенциалов действия, полифазность записи.
Биохимически нередко обнаруживается изменение активности креатиникиназы, АСТ и других ферментов. Иногда обнаруживается изменение состава электролитов крови.
Достоверным считают диагноз при проведении гистологического исследования мышц. Меняются форма и размеры мышечных волокон, изменяется восприятие их окраски гистологическими красителями, мышцы перерождаются, увеличивается объем мышечных ядер. Между мышечными волокнами определяется жир, соединительная ткань. При этом отсутствует пучковость распределение волокон, характерная для нейрогенных миопатий.
Лечение
Патогенетической терапии не разработана. Лечение симптоматическое и направлено на уменьшение скорости прогрессирования. Активно используют витамины группы В, витамин Е, АТФ, экстракт алоэ внутримышечно, АТФ. Некоторое время назад использовался анаболический гормон ретаболил, однако часто отмечалось усиление распада мышечной ткани. Также применяют такие препараты, как тиоктовая кислота, рибоксин, актовегин.
Важная роль отводится немедикаментозным методикам воздействия. Массаж пациентам с дистрофией Эрба-Рота должен проводиться в легком темпе, направлен на борьбу с мышечным спазмом, укрепление мышц. Также важная роль принадлежит ЛФК. ЛФК при заболевании должна быть умеренной, но регулярной, в идеале ежедневной. Тренируются все группы мышц.
Постоянство проведение профилактических мероприятий позволяет длительно сохранять возможность больных к самообслуживанию. В моей практике вспоминается пациентка, которая при дебюте заболевания в 25 лет до 60 сохранила способность к самостоятельному передвижению, самообслуживанию, пускай с ограничением.
Прогноз
Прогноз при всех мышечных дистрофиях, как правило, неблагоприятный. Заболевание постепенно прогрессирует, охватывая все группы мышц. Рано или поздно наступает обездвиживание пациента. Хотя при этом само заболевание практически не приводит к смерти пациента. Смерть обусловлена пролежнями, инфекциями легких, мочевыводящих путей и т.п.
Наследуется по аутосомно-рецессивному типу (болеют дети здоровых родителей).Развитие
: Болезнь начинается в возрасте 14-18 лет. Развивается постепенно атрофия (дистрофия) мышц тазового пояса, приводящая к изменению походки, которая напоминает "утиную". Затем процесс распространяется на мышцы плечевого пояса и мышцы спины.
Симптомы
: Больные испытывают затруднение при вставании с пола, помогая себе при этом руками; по ступенькам лестницы поднимаются, опираясь руками о перила. Слабость и атрофия мышц плечевого пояса затрудняют подъем рук вверх. Атрофия мышц, фиксирующих лопатки, приводит к развитию крыловидных лопаток. Движения же в кистях рук и мышцах лица не нарушаются. Возможно увеличение икроножных мышц голеней, которые становятся плотными на ощупь, однако сила в них снижена. Это псевдогипертрофия икроножных мышц, развивающаяся в результате разрастания в них соединительной и жировой ткани. Интеллект больных не страдает.
Течение заболевания длительное. К 35- 40 годам больные утрачивают способность самостоятельно передвигаться.
ПСЕВДОГИПЕРТРОФИЧЕСКАЯ МИОПАТИЯ ДЮШЕННА
Самая злокачественная из всех болезней нервно-мышечной системы.Это заболевание наблюдается только у мальчиков, т. е. наследуется сцеплено с половой хромосомой. Передается болезнь по материнской линии.
Развитие : Начинается заболевание в детстве, в возрасте 3-5 лет, атрофический процесс захватывает мышцы тазового пояса и бедер, вследствие чего рано появляются затруднения при ходьбе в гору, по лестнице, походка становится утиной. Ребенок часто падает, поднимается с трудом. Изменяется статика. К 10-12 годам больные миопатией Дюшенна теряют способность самостоятельно передвигаться и оказываются прикованными к постели.
При этой форме миопатии может страдать интеллект. Находят изменения и в мышце сердца. Развиваются псевдогипертрофия икроножных мышц. Основу всех прогрессирующих мышечных дистрофий составляет постепенная гибель мышечных волокон скелетной мускулатуры и замена их соединительной и жировой тканью. Вследствие этого развиваются псевдогипертрофия мышц, чаще икроножных (при миопатиях Дюшенна и Эрба) и ретракции (укорочения) ахилловых сухожилий. Гибель мышц происходит в результате патологически измененного обмена веществ о них. Грубо нарушается белковый и углеводный обмен.
Лечение : Диета, богатая белками (рыба, мясо, творог) и витаминами. Ограничивать в движениях не следует, напротив, показаны лечебная гимнастика и массаж. Курсами проводят инъекции ретаболила (по I инъекции в неделю, всего 4 инъекции), АТФ (15-20 инъекций на курс), церебролизип (по 1 мл внутримышечно, 20-30 инъекций), а также анаприлин (обзидан) по 20- 40 мг 2 раза в день (в течение 4 нед. с последующей отменой препарата при ежедневном снижении дозы на 10 мг). Рекомендуется также прием глютаминовой кислоты (по 0,5 г 3 раза в день), ексазила (по 1 таблетке 2 раза в день). Можно проводить дробные переливания одногруппной крови по 250 мл. Курсы лечения длятся 4-6 нед. с перерывом 3-5-12 мес. в зависимости от течения болезни.
ЛОПАТОЧНО-ПЛЕЧЕВАЯ ПРОГРЕССИРУЮЩАЯ МЫШЕЧНАЯ ДИСТРОФИЯ ЛАНДУЗИ-ДЕЖЕРИНА
Впервые описана Landouzy и Dejerine в 1885 году. Заболевание является вторым по распространенности среди прогрессирующих мышечных дистрофий и встречается с частотой 1: 20 000 человек.Виды:
1) классический юношеский 1А тип (ОMIM: 158900);
2) ранний детский;
3) лопаточно-плечевой без вовлечения мышц лица (ОMIM: 600416).
Симптомы и Развитие:
1) Возраст начала классического варианта заболевания варьирует - от 10 до 20 лет. Первые признаки мышечной слабости возникают в мимической мускулатуре, придавая лицу больного специфический маскообразный вид ("лицо сфинкса"). Губы больных утолщаются и выпячиваются ("губы тапира"), возникают трудности при наморщивании лба, складывании губ в трубочку, свисте. Часто при вовлечении в процесс круговой мышцы глаза больные не могут плотно сомкнуть веки, что наиболее заметно во время сна.
Бульбарные, экстрокулярные и дыхательные мышцы, как правило, не поражаются.
У некоторых больных отмечена врожденная аплазия части или целой мышцы (например m.pectoralis) этиология которой не ясна. Предполагается также, что первые признаки мышечной слабости могут быть отмечены в мышцах живота, на которую редко обращают внимание как больные, так и врачи.
Наряду с поражение лицевой мускулатуры на ранних стадиях заболевания отмечается слабость мышц плечевого пояса. Наиболее пораженными оказываются трапецевидные, ромбовидные, грудные и широчайшая мышца спины. Это приводит к ограничению объема движений в плечевом суставе (возникают трудности при подъеме рук выше горизонтального уровня), и появлению так называемых "крыловидных лопаток". Вовлечение в процесс мышц тазового пояса и проксимальных групп мышц ног наблюдается лишь у небольшого процента больных и только в поздних стадиях заболевания (как правило, спустя 15-20 лет от появления первых признаков болезни). На нижних конечностях наиболее пораженными оказываются подвздошные, ягодичные и приводящие мышцы бедер. Характерным признаком заболевания является асимметрия поражения, которая может наблюдаться даже в пределах одной мышечной группы. Псеводогипертрофии мышц не характерны, но могут появлятся в поздней стадии заболевания.
Описано также наличие больных с сочетанием симптомов ЛЛП ПМД с нейросенсорной тугоухостью и аномалией капилляров сетчатки глаза.
Заболевание прогрессирует медленно, не приводя к выраженной инвалидизации и снижению продолжительности жизни и фертильности больных.
2) Раннедетская форма заболевания возникает в возрасте от 3 до 6 лет и быстро прогрессирует, приводя к обездвиженности к 15-20-летнему возрасту.
Клинические проявления сходны с таковыми при классическом варианте заболевания. Для этой формы характерно возникновение контрактур, псевдогипертрофий мышц, а также сколиоза в грудопоясничном отделе.
Описаны больные с ранним началом заболевания в сочетании с олигофренией, эпилепсией, нейросенсоной глухотой и патологией сосудов сетчатки.
Наиболее веротно, что этот клинический вариант не является самостоятельной нозологической формой, так как описано сочетание юношеского и раннедетского варианта в одной и той же семье. Предполагается, что классическая и раннедетская формы болезни являются аллельным вариантами лице-плече-лопаточной мышечной дистрофии.
3) Третий вариант представляет собой редкую форму лопаточно-плечевой прогрессирующей мышечной дистрофии, протекающую без поражения лицевой мускулатуры. Заболевание возникает в возрасте 12-40 лет, характеризуется умеренным прогрессированием и наследуется по аутосомно-доминантному типу.