Полная трисомия 7 хромосомы. Смотреть что такое "7-я хромосома человека" в других словарях
Читайте также
Особую группу заболеваний, связанных со структурными изменениями в генетическом материале» составляют хромосомные болезни, условно относящиеся к категории наследственных. Дело в том, что в подавляющем большинстве случаев хромосомные болезни не передаются потомству, поскольку их носители чаще всего бывают бесплодными.
Хромосомные болезни обусловлены геномнымиили хромосомными мутациями, произошедшими в гамете одного из родителей, или в зиготе, сформированной гаметами с нормальным набором хромосом. В первом случае все клетки будущего ребенка будут содержать аномальный хромосомный набор (полная форма хромосомной болезни), во втором – развивается мозаичный организм, лишь часть клеток которого с аномальным набором хромосом (мозаичная форма болезни). Степень выраженности патологических признаков при мозаичной форме болезни слабее, нежели при полной.
Фенотипическую основу хромосомных болезней составляют нарушения раннего эмбриогенеза, вследствие чего болезнь всегда характеризуется множественными пороками развития.
Частота хромосомных нарушений достаточно высока: из каждой 1000 живорожденных младенцев 3-4 имеют хромосомные болезни, у мертворожденных детей они составляют 6%; дисбалансом хромосом обусловлено около 40% спонтанных абортов (Н.П.Бочков, 1984). Количество вариантов хромосомных болезней не столь велико, как можно было бы ожидать теоретически. Дисбаланс, затрагивающий все пары хромосом, вызывает настолько значительные нарушения в организме, что они, как правило, оказываются несовместимыми с жизнью уже на ранних или более поздних этапах эмбриогенеза. Так, моноплоидия не обнаружена ни у новорожденных, ни у абортусов. Описаны редкие случаи триплаидии и тетраплоидии у абортусов и у живорожденных, которые, однако, погибали в первые дни жизни. Чаще встречаются изменения числа или структуры отдельных хромосом. Недостаток генетического материала вызывает более значительные дефекты, чем избыток. Полные моносомии, например, по аутосомам практически не обнаружены. По-видимому такой дисбаланс вызывает летальный исход уже в гаметогенезе или на стадии зиготы и ранней бластулы.
Основа для развития хромосомных болезней, связанных с изменением числа хромосом формируется в гаметогенезе, во время первого или второго мейотических делений или в период дробления оплодотворенной яйцеклетки, чаще всего в результате нерасхождения хромосом. При этом одна из гамет вместо одинарного набора хромосом содержит крайне редко – диплоидный набор всех хромосом, или 2 хромосомы какой-либо из пар хромосом, вторая гамета не содержит ни одной такой хромосомы. При оплодотворении аномальной яйцеклетки сперматозоидом с нормальным набором хромосом или нормальной яйцеклетки аномальным сперматозоидом, реже при сочетании двух гамет, содержащих измененное число хромосом, создают предпосылки для развития хромосомной болезни.
Вероятность такого рода нарушений, а, следовательно, и рождения детей с хромосомными болезнями, нарастает с возрастом родителей, особенно матери. Так, частота нерасхождения 21-ой пары хромосом в 1-м мейотическом делении составляет 80% всех его случаев, из них в 66,2% – у матери и в 13,8% – у отца; суммарный риск иметь ребенка с трисомией по 13-ой, 18-ой, 21-ой хромосоме для женщины в возрасте 45 лет и старше в 60 раз выше риска для женщины 19-24 лет (Н.П. Бочков и др. 1984).
Самой частой хромосомной болезнью является болезнь Дауна. Кариотип больных в 94% состоит из 47 хромосом за счет трисомии по 21 хромосоме. Примерно в 4% случаев отмечается транслокация лишней 21-ой хромосомы в 14-ю или 22-ю, общее число хромосом равно 46. Болезнь характеризуется резкой задержкой и нарушением физического и психического развития ребенка. Такие дети низкорослы, поздно начинают ходить, говорить. Бросаются в глаза внешний вид ребенка (характерная форма головы со скошенным затылком, широкая, глубоко запавшая переносица, монголоидный разрез глаз, открытый рот, неправильный рост зубов, макроглоссия, мышечная гипотония с разболтанностью суставов, брахидактилия, особенно мизинца, поперечная складка на ладони и др.) и выраженная умственная отсталость, иногда до полной идиотии. Нарушения отмечаются во всех системах и органах. Особенно часты пороки развития нервной (в 67%), сердечно-сосудистой (64,7%) системы. Как правило, изменены реакции гуморального и клеточного иммунитета, страдает система репарации поврежденной ДНК. С этим связана повышенная восприимчивость к инфекции, более высокий процент развития злокачественных новообразований, в особенности лейкозов. В большинстве случаев больные бесплодны. Однако, встречаются случаи рождения больной женщиной детей, часть из них страдают той же болезнью.
Второй по частоте (1:5000-7000 родов) патологией обусловленной изменением числа аутосом, является синдром Патау (трисомия 13). Синдром характеризуется тяжелыми пороками головного мозга и лица (дефекты строения костей мозгового и лицевого черепа, головного мозга, глаз; микроцефалия, расщелина верхней губы и неба), полидактилией (чаще – гексодактилия), дефектами перегородок сердца, незавешенным поворотом кишечника, поликистозом почек, пороками развития других органов. 90% детей родившихся с этой патологией, погибают в течение 1-го года жизни.
Третье место (1:7000 рождений) среди полисемии аутосом занимает трисомия 18 (синдром Эдвардса). Основные клинические проявления болезни: многочисленные пороки костной системы (патология строения лицевой части черепа: микрогнатия, эпикант, птоз, гипертелоризм) сердечно-сосудистой (дефекты межжелудочковой перегородки, пороки клапанов легочной артерии, аорты), гипоплазия ногтей, подковообразная почка, крипторхизм у мальчиков. 90% больных погибает на первом году жизни.
Намного чаще встречаются хромосомные болезни, связанные с нерасхождением половых хромосом. Известные варианты гоносомных полисомий приведены в таблице.
Типы гоносомных полисомий, обнаруженных у новорожденных
(по Н.П.Бочкову, А.Ф. Захарову, В.И.Иванову, 1984)
Как следует из таблицы, подавляющее число полисимий по половым хромосомам приходится на трисомии XXX, XXV, XVV.
При трисомии по Х-хромосоме («сверхженщина») клинические признаки болезни нередко отсутствуют или минимальны. Болезнь диагносцируется по обнаружению вместо одного двух телец Барра и по кариотипу 47,XXX. В других случаях у больных отмечается гипоплазия яичников, матки, бесплодие, различные степени умственной неполноценности. Увеличение в кариотипе числа Х-хромосом увеличивает проявление умственной отсталости. Такие женщины чаще, чем в общей популяции страдают шизофренией.
Варианты полисомий с участием У-хромосом более многочислены и многообразны. Наиболее частый из них – синдром Клайнфельтера – обусловлен увеличением общего числа хромосом до 47 за счет Х-хромосомы. Больной мужчина (наличие У-хромосомы доминирует при любом количестве Х-хромосом) отличается высоким ростом, женским типом строения скелета, инертностью и умственной отсталостью. Генетический дисбаланс обычно начинает проявляться в период полового созревания, недоразвитием мужских половых признаков. Яички уменьшены в размерах, наблюдается аспермия или олигоспермия, часто гинекомастия. Надежным диагностическим признаком синдрома служит обнаружение в клетках мужского организма полового хроматина. Синдром сверхклайн-фельтера (ХХХУ, два тельца Барра), характеризуется большей выраженностью названных признаков, умственная несостоятельность достигает степени идиотии.
Обладатель кариотипа 47, ХУУ – «супер мужчина» отличается импульсивным поведением с выраженными элементами агрессивности. Большое число таких индивидов выявляется среди заключенных.
Гоносомная моносомия встречается намного реже, чем полисомия, и ограничивается лишь моносомией Х (синдром Шерешевского-Тернера). Кариотип состоит из 45 хромосом, половой хроматин отсутствует. Больные (женщины) отличаются низким ростом, короткой шеей, шейными боковыми кожными складками. Характерны лимфатический отек стоп, слабое развитие половых признаков, отсутствие гонад, гипоплазия матки и фолопиевых труб, первичная аменорея. Такие женщины бесплодны. Умственная способность, как правило, не страдает.
Случаев моносомии У не выявлено. По-видимому отсутствие Х-хро-мосомы несовместимо с жизнью и особи типа «ОУ» гибнут на ранних этапах эмбриогенеза.
Хромосомные болезни, обусловленные структурными изменениями хромосом, встречаются реже и, как правило, приводят к более тяжелым последствиям: спонтанным абортам, недоношенности, мертворождению, ранней детской смертности.
На постимплантационных стадиях не зарегистрировано ни одного плода с трисомией хромосом 1 или 19. Предполагается, что трисомия по этим хромосомам вообще не совместима с постимплантационным развитием Однако единичные случаи трисомии 1 обнаружены среди дробящихся зародышей, то есть их развитие возможно, по крайней мере, до стадии 10 бластомеров . Один случай мозаичной трисомии 1 в клетках цитотрофобласта зарегистрирован и в наших исследованиях. По-видимому, на более поздних стадиях такие зародыши либо погибают, либо происходит элиминация бластомеров с дисбалансом этих хромосом.
Трисомия 2 (Тс2) описана лишь у спонтанных абортусов. При этом считается, что Тс2 характерна для клеток мезенхимной стромы ворсин хориона и выявляется лишь на препаратах культивированных клеток хориона . Однако нами выявлен случай Тс2 в цитотрофобласте при развивающейся беременности (табл. 5.5), а в литературе описаны случаи пренатальной диагностики и живорождения детей с мозаичной формой Тс2 .
Тс3 - одна из наиболее частых трисомий, характерных для клеток цитотрофобласта (8 случаев в нашем исследовании), причем доля три- сомных клеток может варьировать от единичных находок до полной формы .
По-видимому, трисомии хромосом группы В, как и большинства хромосом группы С, также летальны и достаточно редки даже в клетках хориона. В наших исследованиях зарегистрирован один случай полной формы трисомии 4, ограниченной цитотрофобластом.
Особого внимания заслуживают хромосомы 7, 8 и 9, для которых отмечена несколько повышенная, по сравнению с остальными хромосомами группы С, частота соответствующих трисомий в материале спонтанных абортов. Случаи Тс7, Тс8 и Тс9, выявленные пренатально и у новорожденных, свидетельствуют о сублетальном эффекте избытка генетического материала этих хромосом. Следовательно, наличие даже мозаичной формы этих трисомий в клетках хориона требует исследования кариотипа плода. Известно, что Тс7 является одной из характерных для трофобласта трисомий (19 случаев в наших исследованиях). Между тем, мозаичные формы трисомии 7 описаны и в культурах клеток амниотической жидкости, а также в фибробластах кожи у детей после рождения . Поэтому мнение, что Тс7 всегда ограничена цитотрофобластом , требует корректировки. Ограниченные плацентой полные формы трисомии по хромосомам группы С
Таблица 5.5. Частота (%) и спектр хромосомных аномалий на разных стадиях онтогенеза
|
Хромосома |
Собственные данные (результаты пренатальной диагностики) N = 7579 |
Данные литературы |
||||
|
Развива ющаяся беремен ность |
Мозаи- цизм, ограниченный плацентой |
Я а о Н VQ нн О о Н о § |
Мертво рожденные |
Живорож денные |
Прогноз жизнеспо собности |
|
|
i |
- |
0,01 |
- |
- |
- |
- |
|
2 |
- |
0,01 |
1,1 |
- |
- |
0 |
|
3 |
- |
0,11 |
0,3 |
- |
- |
0 |
|
4 |
- |
0,01 |
0,8 |
- |
- |
0 |
|
5 |
- |
- |
0,1 |
- |
- |
0 |
|
6 |
- |
- |
0,3 |
- |
- |
0 |
|
7 |
0,026 |
0,23 |
0,9 |
- |
- |
0 |
|
8 |
- |
0,08 |
0,8 |
- |
- |
0 |
|
9 |
- |
0,05 |
0,7 |
0,1 |
- |
0 |
|
10 |
- |
0,01 |
0,5 |
- |
- |
0 |
|
11 |
- |
- |
0,1 |
- |
- |
0 |
|
12 |
- |
- |
0,2 |
- |
- |
0 |
|
13 |
0,2 |
0,02 |
1,1 |
0,3 |
0,05 |
2,8 |
|
14 |
- |
- |
1,0 |
- |
- |
0 |
|
15 |
- |
0,03 |
1,7 |
- |
- |
0 |
|
16 |
- |
0,05 |
7,5 |
- |
- |
0 |
|
17 |
- |
- |
0,1 |
- |
- |
0 |
|
18 |
0,77 |
0,01 |
1,1 |
1,2 |
0,01 |
5,4 |
|
19 |
- |
- |
- |
- |
- |
0 |
|
20 |
- |
0,05 |
0,6 |
- |
- |
0 |
|
21 |
1,64 |
0,1 |
2,3 |
1,1 |
0,12 |
22,1 |
|
22 |
0,013 |
0,05 |
2,7 |
0,1 |
- |
0 |
|
Мозаичные трисомии |
0,05 |
- |
1,1 |
0,5 |
0,02 |
9,0 |
|
Двойные трисомии |
- |
0,01 |
0,8 |
- |
- |
0 |
|
XXY |
0,19 |
- |
0,2 |
0,4 |
0,05 |
55,3 |
|
XXX |
0,09 |
- |
0,1 |
0,3 |
0,05 |
70,0 |
|
XYY |
0,05 |
- |
- |
- |
0,05 |
100,0 |
|
45,Х |
0,43 |
0,4 |
8,6 |
0,25 |
lt; 0,01 |
0,3 |
|
Полиплоидия |
0,25 |
0,01 |
9,8 |
0,6 |
- |
0 |
|
Структурные |
0,12 |
0,01 |
2,0 |
0,4 |
0,6 |
62,0 |
(особенно 6, 7 и 11, в которых локализованы кластеры импринтирован- ных генов), нуждаются в уточняющей диагностике кариотипа плода и исключении однородительской дисомии.
Из трисомий по хромосомам группы D (13, 14, 15) сублетальной является трисомия 13 (синдром Патау). Интересно отметить, что полные формы этой трисомии встречаются чаще, чем мозаичные, в т. ч. ограниченные плацентой. Летальные трисомии 14 и 15, выявленные в трофобласте, заслуживают внимания с точки зрения однородительской дисомии у плода. Поэтому при наличии в образце хориона клеток с трисомией любой из хромосом группы D необходимo кариотипирова- ние плода по лимфоцитам пуповинной крови .
Тс16 является одной из наиболее частых численных аберраций на ранних стадиях развития (среди спонтанных абортусов ее частота составляет 7,5 %). Интересно, что в нашей выборке был выявлен только один случай полной трисомии 16 и два случая с единичными трисом- ными клетками в цитотрофобласте плаценты. К сожалению, кариотип плода ни в одном случае исследовать не удалось. Однако описанные в литературе случаи Тс16 в клетках амниотической жидкости позволяют предполагать, что, по крайней мере, мозаики с таким нарушением кариотипа могут развиваться до второго триместра беременности.
Случаи Тс17 не выявлены в наших исследованиях. В мозаичном варианте они описаны в амниоцитах II триместра , однако частота их невысока.
Тс18 (синдром Эдвардса) как сублетальная мутация встречается на всех стадиях внутриутробного развития. Как и другие частые сублетальные трисомии, Тс18 в основном представлена полными формами и гораздо реже - мозаичными. В нашем исследовании Тс18 была ограничена плацентой лишь в одном случае, тогда как другие авторы отмечают высокую частоту Тс18 в хорионе .
Тс20 долгое время считалась летальной на ранних эмбриональных стадиях. В настоящее время мозаичные случаи Тс20 выявлены пренатально в разные сроки беременности и у детей. Однако комплекс пороков при Тс20 в специфический синдром не выделен. Интересно, что обычно Тс20 ограничена клетками экстраэмбриональных тканей, а у плода она представлена только в клетках некоторых органов (почки, прямая кишка, пищевод) . Все 4 случая полной и мозаичной Тс20 в нашем исследовании оказались ограничены клетками трофобласта.
Согласно многочисленным наблюдениям, для Тс21 (синдром Дауна) характерной является полная форма. В наших исследованиях мозаичная Тс21 с доминирующей диплоидной линией в цитотрофобласте установлена в 4 случаях. Ни в одном из них диагноз не был подтвержден при исследовании лимфоцитов пуповинной крови плода или периферической крови новорожденного. Однако мы полагаем, что все случаи мозаичной Тс21 в цитотрофобласте нуждаются в дополнительных исследованиях на других клетках (амниоциты, лимфоциты пуповинной крови), так как прогноз для жизнеспособности у плодов с трисомией 21, в отличие от других сублетальных трисомий, как правило, благоприятен (22,1 %) (табл. 5.4).
Известно, что Тс22 существует как самостоятельный синдром Тс22, т. е. является сублетальной . Полная форма Тс22 зарегистрирована нами в цитотрофобласте лишь в одном случае; еще в трех она была представлена мозаичным вариантом. 7-я хромосома человека паука, 7-я хромосома человека органы
7-я хромосо́ма челове́ка - одна из 23 человеческих хромосом. Хромосома содержит более 158 млн пар оснований, что составляет от 5 до 5,5 % всего материала ДНК человеческой клетки. настоящее время считается, что на 7-й хромосоме находятся от 1000 до 1400 генов.
7-я хромосома содержит кластер A генов гомеобокса.
- 1 Гены
- 1.1 Плечо p
- 1.2 Плечо q
- 2 Болезни и расстройства
- 2.1 Хромосомные болезни
- 3 Примечания
Гены
Ниже перечислены некоторые гены, расположенные на 7-й хромосоме:
Плечо p
- ABCA13 - 13-й член A-подсемейства АТФ-связывающих кассетных белков;
- AQP1 - Аквапорин 1;
- C1GALT1 - гликозилтрансфераза;
- CBX3 - хромобокс гомолог 3;
- CCM2 - церебральная кавернозная мальформация 2;
- DFNA5 - глухота, аутосомно-доминантный тип 5;
- GARS - глицил-тРНК-синтетаза;
- IL6 - интерлейкин 6;
- ITGB8 - гликопротеин из надсемейства интегринов (β8);
- NOD1 - Nod-подобный рецептор подсемейства NOD;
- PMS2 - PMS2 англ. postmeiotic segregation increased 2 (S. cerevisiae).
Плечо q
- ABCB1 - P-гликопротеин;
- ASL - аргининосукцинат-лиаза;
- CAV1 - кавеолин 1;
- CCL24 - Chemokine (C-C motif) ligand 24 (scya24);
- CCL26 - Chemokine (C-C motif) ligand 26 (scya26);
- CD36;
- CDK5 - циклин-зависимая киназа 5;
- CGRP-RCP - calcitonin gene-related peptide receptor component protein;
- CFTR - Трасмембранный регулятор муковисцидоза, ATP-binding cassette (sub-family C, member 7);
- CLCN1 - хлоридный канал 1;
- CNTNAP2 - ген, ассоциированный с аутизмом;
- COL1A2 - коллаген, тип I, альфа 2;
- CYLN2 - cytoplasmic linker 2;
- DLD - дигидролипоамидная дегидрогеназа (E3 компонент пируват дегидрогеназного комплекса, 2-oxo-glutarate complex, branched chain keto acid dehydrogenase complex);
- ELN - эластин (надклапанный аортальный стеноз, Williams-Beuren syndrome);
- FOXP2 - Forkhead box protein 2;
- GTF2I - general transcription factor II, i;
- GTF2IRD1 - GTF2I repeat domain containing 1;
- GUSB - бета-глюкуронидаза;
- HSPB1 - heat shock 27kDa protein 1;
- KCNH2 - potassium voltage-gated channel, subfamily H (eag-related), member 2;
- KRIT1 - KRIT1, ankyrin repeat containing;
- LIMK1 - LIM domain kinase 1;
- NOS3 - эндотелиальная синтаза оксида азота
- p47 phox или NCF1 - 47 kDa нейтрофил оксидазный фактор / нейтрофил цитозольный фактор 1;
- PIK3CG - каталитическая субъединица γ фосфатидилинозитол-4,5-бисфосфат-3-киназы (PI3K gamma, p110γ);
- RELN - рилин;
- SBDS - Shwachman-Bodian-Diamond syndrome;
- SH2B2 - адаптерный белок;
- SLC25A13 - solute carrier family 25, member 13 (citrin);
- SLC26A4 - solute carrier family 26, member 4;
- SRI - sorcin;
- TAS2R16 - taste receptor, type 2, member 16;
- TFR2 - рецептор трансферрина 2;
- TPST1 - тирозилпротеин сульфотрансфераза 1;
- VGF - «индуцируемый фактором роста нервов».
Болезни и расстройства
Ниже перечислены некоторые заболевания, связанные с генами 7-й хромосомы, а также гены, дефекты которых вызывают эти заболевания:
- аргининосукциновая ацидурия (англ. argininosuccinic aciduria) - ASL;
- бессиндромная глухота (англ. nonsyndromic deafness) аутосомно-доминантный тип 5 и аутосомно-рецессивный тип 4 - DFNA5 и SLC26A4;
- болезнь мочи с запахом кленового сиропа - DLD;
- болезнь Шарко - Мари - Тута (англ. Charcot–Marie–Tooth disease) типов 2В и 2F - GARS и HSPB1;
- врожденное двустороннее отсутствие семявыносящих протоков (англ. congenital bilateral absence of vas deferens) - CFTR;
- гемохроматоз типа 3 (англ. hemochromatosis, type 3) - TFR2;
- дистальная спинальная амиотрофия (англ. distal spinal muscular atrophy) типа 5 - GARS;
- кавернозная ангиома (англ. cerebral cavernous malformation) - CCM2;
- миелодиспластический синдром;
- муковисцидоз - CFTR;
- мукополисахаридоз типа VII, или синдром Слая - GUSB;
- наследственный неполипозный колоректальный рак (англ. hereditary nonpolyposis colorectal cancer) - PMS2;
- несовершенный остеогенез типов I, II, III и IV - COL1A2;
- сахарный диабет взрослого типа у молодых типа 2 - GCK;
- синдром Вильямса - ASL, BAZ1B, BCL7B, CLDN3, CLDN4, CLIP2, EIF4H, ELN, FZD9, FKBP6, GTF2I, GTF2IRD1, HIP1, KCTD7, LAT2, LIMK1, MDH2, NCF1, NSUN5, POR, RFC2, STX1A, TBL2, TRIM50, TRIM73, TRIM74, WBSCR14, WBSCR18, WBSCR21, WBSCR22, WBSCR23, WBSCR24, WBSCR27 и WBSCR28;
- синдром Норман - Робертс - RELN;
- синдром Пендреда - SLC26A4;
- синдром Романо - Уорда (англ. Romano–Ward syndrome) - KCNH2;
- синдром Швахмана - Даймонда (англ. Shwachman–Diamond syndrome) - SBDS;
- синдром Элерса - Данлоса с артрохалазией типа 7B - COL1A2;
- хроническая гранулематозная болезнь, обусловленная недостаточностью нейтрофильного цитозольного фактора 1 - NCF1
- цитруллинемия (англ. citrullinemia) типа II - SLC25A13;
- шизофрения - KCNH2, ABCA13
Хромосомные болезни
Некоторые расстройства вызываются изменениями в структуре или количестве копий 7-й хромосомы:
- синдром Вильямса - делеция участка длинного плеча хромосомы в позиции 7q11.23, который содержит более 20 генов; потеря некоторых из этих генов и связана с характерными особенностями расстройства, однако для большинства генов удалённого участка связь с симптомами пока не установлена;
- задержка роста и развития, умственная отсталость, характерные изменения черт лица, скелетные аномалии, замедленная речь и другие медицинские проблемы - дополнительная копия части хромосомы (частичная трисомия) или отсутствие части хромосомы (частичная моносомия), иногда делеция или дупликация части хромосомы, а также возникновение кольцевой хромосомы (англ. ring chromosome).
Примечания
- Human chromosome 7 map view (англ.). Vertebrate Genome Annotation (VEGA) database. The Wellcome Trust Sanger Institute. - Карта хромосомы и её основные параметры: размер, количество генов и т. п. Проверено 8 сентября 2009. Архивировано из первоисточника 1 апреля 2012.
- Kratz C. P., Emerling B. M., Bonifas J., Wang W., Green E. D., Le Beau M. M., Shannon K. M. Genomic structure of the PIK3CG gene on chromosome band 7q22 and evaluation as a candidate myeloid tumor suppressor // Blood. - 2002. - Т. 99, вып. 1. - С. 372-374. - PMID 11756194.
Кариотип клеток костного мозга у больных с (МДС) интенсивно изучается на протяжении последних 10-15 лет. Аномальные клоны выявлены до лечения у 30-50 % пациентов, в некоторых сообщениях приведены более высокие показатели - 60-75 %.
Обнаружение клонов клеток с аномальным кариотипом при миелодиспластическом синдроме (МДС) имеет важное теоретическое и клиническое значение, поскольку свидетельствует о принадлежности этой группы заболеваний к новообразованиям.
Цитогенетические изменения весьма разнообразны , спектр их близок к спектру хромосомных аномалий, наблюдаемых при остром нелимфобластном лейкозе, особенно вторичных.
Наиболее характерны моносомии 5 и 7 , а также делеции длинного плеча этих хромосом, появление дополнительной хромосомы 8 и делеции длинного плеча хромосомы 20.
Установлено, что частота обнаружения клонов анеуплоидных клеток нарастает по мере прогрессирования болезни: на относительно ранних этапах она составляет 20-30 %, при появлении начальных признаков трансформации в острый лейкоз - до 40-60 %, при трансформации в острый миелобластный лейкоз - 80-90 %.
Транслокации, специфичные для первичных острых нелимфобластных лейкозах , при миелодисплазиях наблюдаются редко. Есть сообщения о повторяющихся транслокациях t(3;3)(q21;q26), t(8;21)(q22;q22) и t(3;21)(q26;q22). Примеры перестроек длинного плеча хромосомы 3 показаны на рисунке.
Частота (в процентах) характерных аномалий кариотипа при различных миелодиспластических синдромах
Основные хромосомные аномалии, характерные для миелодисплазий
:
-7 или 7q-
-5 или 5q-
t(1;7)(q10;p10)
del(12)(р12-р13)
t(2;ll)(p13;q23)
del(13)(обязательно с включением 3q14)
t(6;9)(p23;q34)
del(20)(q11ql3)
+8
t(1;3)(p36;q21)
Перечисленные хромосомные аномалии наблюдаются при различных формах миелодисплазий, но частота их несколько различается.
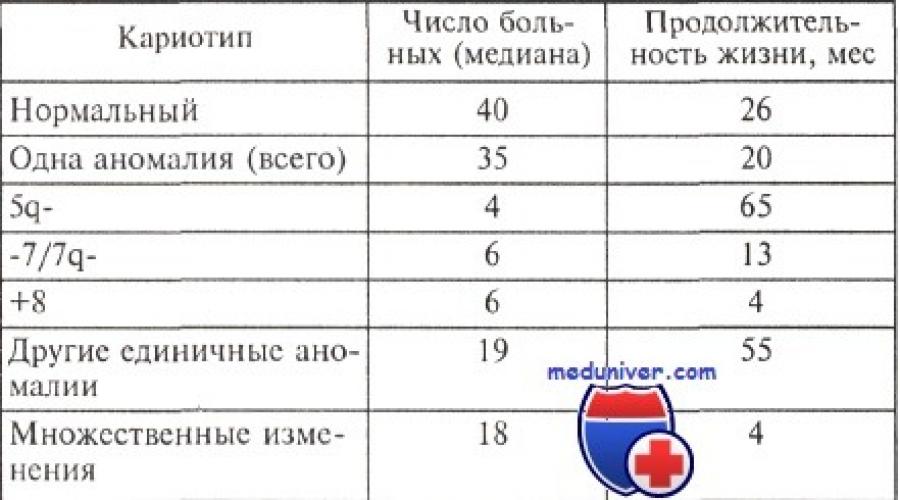
Опыт большинства исследователей свидетельствует о том, что существует корреляция между особенностями кариотипа и продолжительностью жизни больных с миелодиспластическим синдромом (МДС). Относительно благоприятным считается прогноз, если выявлены клоны клеток с единственной перестройкой 5q- или 20q; в то же время при любом варианте миелодиспластического синдрома обнаружение клона с множественными хромосомными аномалиями является крайне неблагоприятным.
Остановимся подробнее на отдельных нарушениях кариотипа, характерных для миелодиспластического синдрома (МДС).
Синдром 5q
- рефрактерная сидеробластная анемия у пожилых больных, преимущественно женщин. В новой классификации ВОЗ этот синдром выделен как самостоятельный вариант миелодиспластического синдрома (МДС). Характерна макроцитарная анемия, резистентная к лечению, в костном мозге - признаки миелодисплазии клеток красного ряда и мегакариоцитов. Число тромбоцитов нормально или повышено, в костном мозге наблюдается гиперплазия гиполобулярных микромегакариоцитов. Клиническое течение сравнительно медленное. Трансформации в острый лейкоз наблюдаются приблизительно в 10 % случаев. Синдром впервые описан van den Berghe и соавт. в 1974-1985 гг..
Делеции длинного плеча хромосомы 5
наблюдаются и при других гематологических заболеваниях.
Предполагают, что делетирующийся участок содержит один или более генов-супрессоров. В этом направлении ведутся интенсивные исследования. До настоящего времени не подтверждена важная роль в патогенезе рефрактерной анемии ни одного из изучавшихся кандидатов на роль гена- супрессора.
Продолжительность жизни больных миелодиспластическим синдромом с различными изменениями кариотипа

Синдром моносомии хромосомы 7 встречается преимущественно у мальчиков до 4 лет. Характерна спленомегалия, часто наблюдаются лейкоцитоз с моноцитозом, тромбоцитопения, анемия. Прогноз плохой.
Как отмечалось, утрата одной из хромосом 7-й пары (моносомия 7) наблюдается при самых различных гемобластозах, включая острый нелимфобластный лейкоз, при этом она обычно ассоциирована с неблагоприятным прогнозом.
Делеции короткого плеча хромосомы 17 (17р-) обычно входят в число сложных изменений кариотипа. Как правило, 17р- сочетается с двумя или более хромосомными аномалиями и имеет неблагопрятное прогностическое значение.
В 75 % случаев в присутствии маркера 17р- наблюдается своеобразный дисгранулоцитопоэз в виде псевдопельгеровских гиподольчатых ядер и вакуолизации цитоплазмы. Этот маркер обнаружен не только при миелодисплазиях, но и при самых разнообразных злокачественных новообразованиях, включая солидные опухоли, его присутствие - плохой прогностический признак.
В 1997 г. опубликованы материалы международного совещания , посвященного диагностике и прогнозированию миелодиспластического синдрома. На основании ретроспективной оценки длительности заболевания до перехода в острый лейкоз и общей продолжительности жизни больных результат цитогенетического анализа был расценен как важнейший прогностический признак. В группу с благоприятным прогнозом отнесены случаи с единичными хромосомными аномалия ми: -Y, 5q- и 20q-. Неблагоприятное течение наблюдали при множественных (сложных) нарушениях (три или более перестройки кариотипа) и изменениях хромосомы 7 (делеции длинного плеча, моносомии).
Другие аномалии определяли «промежуточный» прогноз. Средняя длительность заболевания до перехода в острый лейкоз составила по группам 9,4; 0,4 и 1,1-3,3 года соответственно. Эти данные используются при оценке эффективности новых схем лечения миелодисплазии и зарекомендовали себя как одна из лучших прогностических систем при миелодиспластическом синдроме.
Важное диагностическое значение может иметь метод FISH в тех случаях, когда стандартное цитогенетическое исследование неинформативно или обнаруживаются только единичные клетки с нарушением кариотипа, которое по формальным критериям нельзя считать клоном. Для диагностики наиболее характерных хромосомных нарушений при миелодиспластическом синдроме разрабатывается панель FISH-зондов.
Попытки выделить цитогенетические особенности каждой из клинико-морфологических субъединиц, входящих в общую гетерогенную группу миелодиспластических синдромов, не увенчались успехом. В то же время ХММЛ, рассматриваемый как миелопролиферативное заболевание с морфологическими признаками миелодисплазии, нередко ассоциируется со специфической хромосомной аномалией t(5;12)(q33;p13), однако в большинстве случаев ХММЛ эта хромосомная аномалия не выявляется.