Laborfunktionsanalyse organischer Verbindungen. Physikalisch-chemische Methoden zur Analyse organischer Verbindungen
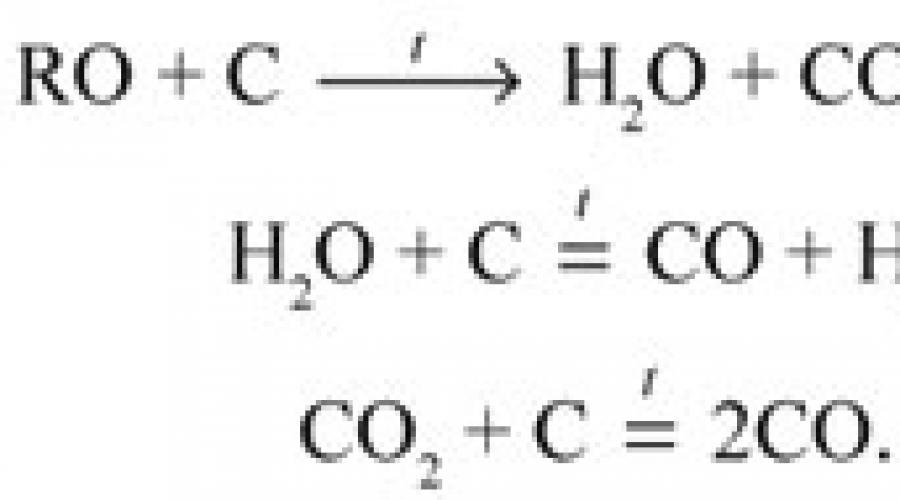
Yu. V. Golubkov,
G. N. Golubkova
Nachweis organischer Substanzen
Für Bewerber an Universitäten, Gymnasiasten, Lyzeen, Gymnasien, Studenten sowie Chemielehrern
Fortsetzung. Siehe den Anfang in Nr. 10/2010
Kapitel II.
Elementaranalyse organischer Stoffe und Bestimmung ihrer Struktur
Gereinigte organische Stoffe werden einer qualitativen und quantitativen Analyse unterzogen. Am häufigsten enthalten organische Verbindungen neben Kohlenstoff und Wasserstoff Sauerstoff, Stickstoff, Schwefel, Halogene und Phosphor.
Die qualitative und quantitative Analyse organischer Verbindungen basiert auf deren Zerstörung (Oxidation), gefolgt von der Bestimmung von CO 2, H 2 O, N 2, HCl usw. mit herkömmlichen Methoden.
Die Elementaranalyse organischer Stoffe begann mit der Bestimmung von Kohlenstoff und Wasserstoff, daher ist es interessant, sich näher mit diesem Thema zu befassen.
§ 7. Analytische Bestimmung von Kohlenstoff und Wasserstoff
Die Essenz der Methode.
Eine abgewogene Menge der zu testenden organischen Verbindung wird mit dem einen oder anderen Oxidationsmittel oxidiert. Kohlenstoffoxid (IV) wird quantitativ aus Kohlenstoff und Wasser aus Wasserstoff gewonnen. Die Masse dieser Verbrennungsprodukte (Oxidation) wird durch das Gewicht bestimmt. Das Verbrennungsrohr (Oxidationsrohr) ist so gefüllt, dass andere in der organischen Verbindung vorhandene Elemente die endgültige quantitative Absorption von Kohlenmonoxid (IV) und Wasser in Absorptionsapparaten mit entsprechenden Absorbern nicht beeinträchtigen.
1) C + O 2 = CO 2,
Kohlendioxid wird von Schwerspatwasser (Bariumhydroxidlösung Ba(OH)2) und Kalkwasser (Kalziumhydroxidlösung Ca(OH)2), Natronkalk (eine Mischung aus 83 % Ca(OH)2, 5 % NaOH und 12) absorbiert % H2O ) und andere Stoffe.
2) H 2 + 1/2O 2 = H 2 O,
Wasser wird durch wasserfreies Magnesiumperchlorat Mg(ClO 4) 2 (Anhydron) absorbiert; In der Intensität seiner Trocknungswirkung kommt es dem Phosphor(V)-oxid nahe und hat gegenüber diesem den wichtigen Vorteil, dass das Magnesiumsalz, das Wasser aufgenommen und durch längeres Erhitzen auf 220 °C im Vakuum geschmolzen wurde, wieder entwässert werden kann wiederverwendet. Es können auch andere Absorber verwendet werden, beispielsweise Calciumchlorid (von dieser Verbindung stammt der Name „Calciumchlorid-Röhrchen“, siehe unten).
Ausrüstung.
Um festzustellen, ob ein bestimmter Stoff zu organischen Verbindungen gehört, muss zunächst festgestellt werden, ob darin Kohlenstoff vorhanden ist. Manchmal bereitet dies keine Schwierigkeiten: Viele organische Stoffe verkohlen beim Erhitzen, d. h. verwandeln sich in Kohle, was auf das Vorhandensein von Kohlenstoff in ihnen hinweist. In einigen Fällen verkohlen kohlenstoffhaltige Stoffe jedoch beim Erhitzen nicht: Wenn Sie beispielsweise Alkohol erhitzen, verdampft dieser einfach, und wenn er sich entzündet, verbrennt er ohne Ruß. Daher besteht der zuverlässigste Weg, Kohlenstoff in einer organischen Verbindung zu entdecken, darin, diese Verbindung vollständig zu verbrennen und das Vorhandensein von Kohlenmonoxid in den Verbrennungsprodukten festzustellen.
Um eine möglichst geringe Menge des Stoffes zu verbrennen und die entstehenden gasförmigen Verbrennungsprodukte nicht zu verlieren, wird eine kleine Menge organischer Stoffe mit einem Oxidationsmittel vermischt und erhitzt. Zu diesem Zweck können keine allzu aktiven Oxidationsmittel verwendet werden, weil Die auftretende Reaktion erschwert das Auffangen der vom Gerät abgegebenen Gase. Wenn man beispielsweise einen brennbaren Stoff mit einem so starken Oxidationsmittel wie Kaliumchlorat KClO 3 (Berthollet-Salz) mischt, dann entsteht bekanntlich beim Erhitzen der Mischung ein Blitz, der an eine Explosion grenzt. Daher werden in diesem Fall niedrigaktive Oxidationsmittel verwendet, die nur bei einem signifikanten Temperaturanstieg wirken, wodurch durch Änderung der Temperatur die Oxidationsrate organischer Stoffe reguliert werden kann. Bei organischen Verbrennungen wird üblicherweise Kupfer(II)-oxid als Oxidationsmittel verwendet.
Für qualitative Definition Nehmen Sie einige Milligramm der Testsubstanz und mischen Sie diese mit einer großen Menge (Überschuss) körnigem Kupfer(II)-oxid, gießen Sie die Mischung in ein Reagenzglas, verschließen Sie es mit einem Stopfen mit Gasauslassröhrchen und erhitzen Sie es. Kupfer(II)-oxid wird durch organische Stoffe reduziert; Durch das Gasauslassrohr austretende Oxidationsprodukte organischer Stoffe werden in ein Gefäß mit einer gesättigten Lösung von Calciumhydroxid (Kalkwasser) geleitet. Das Vorhandensein von Kohlenmonoxid (IV) lässt sich bekanntermaßen anhand der Trübung von Kalkwasser erkennen. Enthält der Verbrennungsstoff Wasserstoff, lagert sich das durch Oxidation entstehende Wasser in Form von Tautropfen an den kalten Teilen des Gerätes ab und kann so nachgewiesen werden.
Um zu produzieren Quantifizierung Kohlenstoff und Wasserstoff müssen Sie die entstehenden Verbrennungsprodukte jeweils einzeln mit etwas absorbieren und wiegen. Das Funktionsprinzip bleibt gleich, lediglich die Ausführungstechnik ändert sich.
Die klassische Verbrennungsmethode, die einst von J. Liebig entwickelt wurde, sieht folgendermaßen aus: Die verbrannte Substanz wird nicht mit Kupfer(II)-oxid vermischt, sondern in ein Porzellan- oder Platinschiffchen eingewogen, das dann in ein spezielles Röhrchen gestellt wird Verbrennung (Abb. 1). Hierbei handelt es sich um ein langes, feuerfestes Glasrohr, in das eine lange Schicht aus körnigem Kupfer(II)-Oxid gegossen wird, das von zwei kleinen Spiralen aus aufgerolltem Kupfergeflecht an Ort und Stelle gehalten wird. Diese Spiralen dringen mit leichter Reibung in das Rohr ein und ihre Oberfläche wird voroxidiert. In das gegenüberliegende Ende des Rohres wird eine längere Spirale aus oxidiertem Kupfergeflecht eingeführt, so dass zwischen ihr und der Schicht aus Kupfer(II)-Oxid ein Spalt verbleibt, der länger ist als bei einem Porzellanschiffchen mit einer suspendierten Substanz.
Das so vorbereitete Rohr wird in einen Verbrennungsofen gestellt und seine Enden werden mithilfe von Gummistopfen mit darin eingesetzten Glasröhrchen mit zuvor gewogenen Absorptionsvorrichtungen verbunden: dem Ende des Rohrs, in dem sich die Kupfer(II)-Oxidschicht befindet befindet sich mit einem Calciumchloridrohr (Abb. 2), in dem der bei der Verbrennung des verbrannten Stoffes entstehende Wasserdampf absorbiert wird, und danach mit einem Kaliumapparat, der mit einer konzentrierten Kaliumhydroxidlösung gefüllt ist und Kohlenmonoxid absorbieren soll ( IV) wird platziert. In Abb. Abbildung 3 zeigt die verschiedenen Ausführungen der verwendeten Kaliapparate. Das gegenüberliegende Ende des Verbrennungsrohrs ist mit Gasometern verbunden, die gefüllt sind: eines mit Luft, das andere mit Sauerstoff (sie werden abwechselnd verwendet). Zwischen den Gasometern und dem Rohr sind außerdem Absorptionsvorrichtungen angebracht, um Feuchtigkeit und Kohlenmonoxid (IV) zurückzuhalten, die möglicherweise in den Gasen enthalten sind, die die Gasometer füllen.
Bestimmungsmethode.
Wenn das Gerät auf diese Weise zusammengebaut wird, wird der mit Kupfer(II)-Oxid gefüllte Teil der Röhre auf eine dunkelrote Hitze erhitzt, dann wird die Kupferspirale aus dem gegenüberliegenden Teil der Röhre, einem Schiffchen mit der Substanz, entfernt Wird eingeführt und drückt die Spirale wieder hinein, verschließt sich mit einem Stopfen und stellt so eine Verbindung zum mit Luft gefüllten Gasometer her. Danach beginnen sie, die lange Spirale zu erhitzen und erhöhen dann vorsichtig die Temperatur des Teils des Rohrs, in dem sich das Schiffchen mit der Substanz befindet, indem sie sehr langsam einen Luftstrom durch das Rohr leiten.
Die Substanz im Boot verdampft teilweise und zersetzt sich teilweise unter Bildung flüchtiger Substanzen. Diese brennbaren Dämpfe, die sich mit Luft vermischen, werden durch die Strömung ihres Strahls zum Ausgang des Rohrs transportiert und bei Kontakt mit heißem Kupfer(II)-oxid oxidiert, wodurch Kupfer(II)-oxid zu Kupfermetall reduziert wird. Wenn Die Oxidation erfolgt zu schnell und zu viele ihrer Produkte (dies lässt sich anhand der Anzahl der durch den Kaliapparat strömenden Blasen beurteilen) senken die Temperatur und verringern die Intensität des Luftstroms; andernfalls bewirken sie das Gegenteil.
Verdampft der zu verbrennende Stoff vollständig oder zersetzt er sich, ohne dass sich Kohle bildet, ist am Ende der Verbrennung nichts mehr im Boot übrig. Wenn sich im Boot Kohle bildet, wird dieser Teil des Rohrs stärker erhitzt und anstelle von Luft beginnt Sauerstoff durchzuströmen, in dem die Kohle nach und nach verbrennt.
Wenn die Verbrennung abgeschlossen ist, wird erneut Luft durch das Rohr geleitet, bis der Sauerstoff, der das Rohr und die Absorptionsvorrichtungen füllt, durch Luft verdrängt wird. Dies ist erforderlich, da die Absorptionsvorrichtungen beim Wiegen vor dem Brennen mit Luft und nicht mit Sauerstoff gefüllt waren. Anschließend werden das Calciumchloridrohr und der Messschieber gewogen, damit sie die Temperatur der Waage erreichen. Die Gewichtszunahme des Calciumchloridröhrchens zeigt die Menge des absorbierten Wassers an, und die Gewichtszunahme des Calciumchloridröhrchens zeigt die Menge des absorbierten Kohlenmonoxids (IV).
Liebig führte seine Analysen mit Holzkohle durch: Das Verbrennungsrohr wurde in eine Wanne gelegt und mit einer Schicht brennender Kohle bedeckt. Um die Temperatur an einer Stelle des Rohres zu senken, wurde die Kohle abgeharkt; Um die Temperatur zu erhöhen, wurde er mit handgefertigten Blasebälgen aufgeblasen. Mit der Einführung von Leuchtgas in die Laborpraxis wurden Öfen mit einer großen Anzahl von Gasbrennern eingesetzt (Abb. 4), deren Flamme leicht zu regulieren ist. In der modernen Praxis werden immer häufiger Elektroöfen eingesetzt.
Liebig-Methode der organischen Analyse, auch genannt elementare Analyse(Bestimmung der elementaren Zusammensetzung organischer Stoffe) wird in Laboratorien immer noch häufig eingesetzt. Zusätzlich zu dieser Methode werden häufig andere verwendet. Beispielsweise erfolgt die Verbrennung nach dem Denstedt-Verfahren ohne Kupfer(II)-oxid; Die Substanz wird in einem Sauerstoffstrom in Gegenwart von Platinmetall verbrannt, das als Katalysator fungiert. Dadurch ist es möglich, die Rohrgröße zu verringern und die Anzahl der Brenner im Ofen zu reduzieren.
In den letzten Jahren hat sich die vereinfachte Verbrennungsmethode von G. Ter-Meulen und I. Gesling immer weiter verbreitet, die darin besteht, den Analyten in einem Sauerstoffstrom zu verbrennen, wobei Mangan(IV)-oxid als Katalysator verwendet wird; Der Ofen, in dem die Verbrennung durchgeführt wird, ist sehr einfach aufgebaut und wird mit nur zwei Brennern beheizt. Diese Methode ist auch insofern praktisch, als sie die Verwendung sehr kleiner Stoffmengen für die Verbrennung ermöglicht und ist Halbmikro-Methode.
Aktuell in Verwendung Mikromethoden organische Analyse. Die besondere Bauweise von Mikrowaagen ermöglicht das Abwiegen kleinster Mengen einer Substanz mit einer Genauigkeit von 0,001 mg. Das Brennrohr und die Absorptionseinrichtungen wurden entsprechend verkleinert und verbessert.
Die ursprüngliche Methode der organischen Mikroanalyse wurde von M. O. Korshun und V. A. Klimova entwickelt. Sein Wesen liegt darin, dass die Verbrennung in einem leeren Rohr im Sauerstoffstrom erfolgt. Eine Probe von 3–5 mg der Substanz wird in ein zylindrisches Quarzglas gegeben, das in ein Verbrennungsrohr (in horizontaler Position) eingeführt wird; Durch das Rohr wird ein Sauerstoffstrom geleitet. Wenn der Abschnitt des Rohrs, in dem sich der Becher mit der brennbaren Substanz befindet, erhitzt wird, kommt es im Inneren des Bechers zu einer Zersetzung und Verkohlung der Substanz, weil Sauerstoff kann im Becher nicht zirkulieren und ist daher nicht im Übermaß vorhanden. Die Produkte der thermischen Zersetzung der analysierten Substanz, die den Becher in ein auf 850–950 °C erhitztes Verbrennungsrohr verlassen, treffen auf einen großen Sauerstoffüberschuss und verbrennen vollständig. Auch die restliche Holzkohle im Becher verbrennt. Das Brennen dauert nur 10–15 Minuten. Somit ist die Mikromethode von Korshun und Klimova eine Hochgeschwindigkeitsmethode.
Wenn es verbrannt war A g Substanz, Gewichtszunahme von Calciumchlorid-Röhrchen – R g (gebildete Wassermasse) und die Gewichtszunahme des Kaliapparats beträgt R 1 g (die Masse des gebildeten Kohlenmonoxids (IV), der prozentuale Anteil an Wasserstoff und Kohlenstoff in der Testsubstanz wird wie folgt bestimmt. 1/9 der Masse des Wassers ist Wasserstoff, also in R g Wasser enthält ( R/9) g Wasserstoff; Um diesen Betrag in Prozent auszudrücken, multiplizieren Sie ihn mit 100 und dividieren ihn durch das Gewicht: R 100/(9A)%. In einem Kohlenmonoxid(IV)-Molekül sind 12/44 seiner Masse Kohlenstoff, daher enthält diese Probe (12 R 1/44) g Kohlenstoff, also 12 R 1 100/(44A) %.
Wenn der zu verbrennende Stoff aus Kohlenstoff, Wasserstoff und Sauerstoff bestand, wird die Masse des letzteren durch die Differenz ermittelt (von 100 % wird die Summe der Massenanteile von Wasserstoff und Kohlenstoff in Prozent abgezogen).
Bei der Bestimmung von Kohlenstoff und Wasserstoff nach der Unterzaucher-Methode wird der Analyt über Kupfer(II)-oxid in einem Luftstrom verbrannt. Wasserstoff wird durch die Menge an freigesetztem H 2 O bestimmt, das mit wasserfreiem BaCl 2 gebunden ist, und Kohlenstoff wird durch die Menge an CO bestimmt, die entsteht, wenn das freigesetzte CO 2 über auf 1120 °C erhitzte Kohle geleitet wird. In diesem Fall wird der CO-Gehalt wie unten beschrieben bestimmt (siehe § 10). Die Methode wurde 1950 von J. Unterzaucher entwickelt.
In der Literatur werden viele weitere Möglichkeiten zur instrumentellen Gestaltung von Methoden zur Bestimmung von Kohlenstoff und Wasserstoff in organischen Verbindungen beschrieben. Achten wir auf die Gründlichkeit der Methoden.
§ 8. Analytische Bestimmung von Halogenen
Halogen liegt in organischen Stoffen nicht in Form eines Ions vor und kann daher nicht mit Silbernitrat in Form von AgCl, AgBr oder AgI ausgefällt werden. Um das Vorhandensein eines Halogens in einer organischen Substanz zu entdecken, muss diese zerstört werden und dabei das Halogen freisetzen oder in eine andere Verbindung umwandeln, in der das Halogen leicht zu öffnen ist.
Ein qualitativer Test auf Halogen kann wie folgt durchgeführt werden: Die Prüfsubstanz wird auf einem oxidierten Kupferdraht oder auf einem Stück Kupfer(II)-oxid, das in der Öse eines Kupferdrahtes befestigt ist, gesammelt und ins Feuer gelegt; Organisches Material verbrennt und erzeugt Kupferhalogenid, das die Flamme grün oder bläulichgrün färbt. Diese Methode zum Öffnen eines Halogens ist bekannt als Beilstein-Tests. Auf diese Weise können Sie das Vorhandensein eines Halogens feststellen, aber Sie können nicht entscheiden, welches davon Teil der untersuchten Substanz war. Darüber hinaus ist die Reaktion nicht selektiv (Nitril, einige Pyridinderivate usw. stören).
Gravimetrische Methoden.
Um Halogen in organischen Verbindungen quantitativ zu bestimmen, können Sie wie folgt vorgehen: Eine Probe der Prüfsubstanz, etwas Silbernitrat und einige Milliliter konzentrierte Salpetersäure werden in ein dickwandiges, an einem Ende verschlossenes Glasröhrchen gegeben. Das Röhrchen wird versiegelt und in einem speziellen Ofen auf 280–300 °C erhitzt, nachdem es zuvor in ein Metallgehäuse gelegt wurde, für den Fall, dass das Röhrchen platzt und dem Druck nicht standhalten kann. Unter diesen Bedingungen verbrennt die organische Substanz (Salpetersäure ist ein Oxidationsmittel) und es bildet sich im Rohr Silberchlorid, -bromid oder -iodid, je nachdem, welches Halogen in der untersuchten Substanz enthalten ist. Durch die Bildung von Kohlenmonoxid (IV) und Stickoxiden entsteht im Rohr ein hoher Druck. Nach dem Abkühlen wird das Röhrchen geöffnet, der Niederschlag entnommen, gewaschen, getrocknet und gewogen. Das - „Nassbrennmethode“ von L. Carius(1860).
Die quantitative Bestimmung von Halogen in alkohollöslichen Halogenderivaten wurde durch A. V. Stepanov (1906) erheblich vereinfacht. Metallisches Natrium wird Stück für Stück zu einer alkoholischen Lösung einer abgewogenen Portion des Halogenderivats gegeben und in einem Kolben unter Rückfluss erhitzt. Unter dem Einfluss von Wasserstoff, der durch Natrium aus Alkohol verdrängt wird, entstehen der entsprechende Kohlenwasserstoff und Halogenwasserstoff, der mit Natrium das entsprechende Salz, beispielsweise Natriumchlorid, ergibt. Dadurch geht das Halogen in einen ionischen Zustand über und wird mit herkömmlichen analytischen Methoden (gravimetrisch oder titrimetrisch) bestimmt.
Bei der Verbrennung von halogenhaltigen Stoffen zur Bestimmung von Kohlenstoff und Wasserstoff wird am Ende des Brennrohrs eine Spirale aus Silbergeflecht angebracht. Der Zweck der Spirale besteht darin, das bei der Verbrennung des Stoffes freigesetzte Halogen zurückzuhalten, damit es zusammen mit Kohlenmonoxid (IV) nicht in den Kalyapparat (Absorptionskolben) eindringt und nicht vom Alkali absorbiert wird.
Von Methode „Trockenbrennen“. Der Stoff wird in einem Quarzrohr einer speziellen Verbrennungsvorrichtung im Sauerstoffstrom verbrannt und die Zersetzungsprodukte werden über erhitzte Platinkontakte geleitet, um eine vollständige Oxidation zu gewährleisten.
Von „Bomben“-Fusionsmethode Die organische Substanz, die das Halogen enthält, wird in einer „Bombe“ mit einer Mischung aus Kaliumnitrat, Natriumperoxid und Rohrzucker (Saccharose) verschmolzen. Dabei entsteht Natriumhalogenid, das mit Silbernitrat ausgefällt und als Silberhalogenid suspendiert wird. Die zur Fusion verwendete „Bombe“ (Abb. 5) besteht aus einem Metallreagenzglas mit einer Tiefe von 25 mm und einem Innendurchmesser von 13 mm; Es besteht aus einer Legierung aus Chrom und Nickel. Die Dicke der Rohrwände beträgt 1,5 mm; Es verfügt über einen 3 mm breiten Flansch, auf den der Deckel und die Bleidichtung aufgesetzt werden. Der runde Boden des Reagenzglases ist mit einer kleinen Öse versehen. Die „Bomben“-Abdeckung wird mit einer bogenförmigen Klemme mit fest sitzender Schraube fest befestigt.
Durchführen einer Analyse
Mischen: 300 mg einer Mischung aus Kaliumnitrat und Rohrzucker (im Verhältnis 3:1) und 1,5 g Natriumperoxid in eine Wägeflasche einwiegen. Ein Drittel dieser Mischung wird in eine „Bombe“ geladen und mit der entsprechenden Menge des Analyten versetzt. Dann wird die restliche Mischung hinzugefügt, der Deckel der „Bombe“ fest aufgeschraubt und der Inhalt durch Schütteln gründlich vermischt. Dieses gründliche Mischen ist sehr wichtig, um eine vollständige Oxidation zu erreichen. Durch wiederholtes Klopfen sammelt sich der Inhalt der „Bombe“ am Boden – die Reaktionsmischung ist zur Fusion bereit.
Fusion. Die „Bombe“ wird vom Kopf einer Schraubzwinge mit einer Zange oder einem Stück starken Drahtes gehalten, der in das Loch im Kopf der Schraube eingeführt wird, und nach und nach zu einem Drittel in den oberen Teil der kleinen farblosen Flamme abgesenkt eines Gasbrenners (Heizung sollte vermieden werden, „Bomben“ befindet sich zu nahe am Deckel). Die Fusion endet in 10 s; Die Fusion selbst ist leicht am Zittern der „Bombe“ zu erkennen. Theoretisch kann die Fusion nach den angegebenen 10 s als abgeschlossen betrachtet werden, es wird jedoch empfohlen, die „Bombe“ weitere 5 s in der Flamme zu belassen, um eine vollständige Fusion des gesamten Reaktionsgemisches sicherzustellen. Anschließend wird die „Bombe“ mit Leitungswasser abgekühlt.
Kühlen und Filtern: Zunächst wird die Außenseite der „Bombe“ mit destilliertem Wasser gewaschen. Dann wird es geöffnet und die Innenseite des Deckels mit heißem destilliertem Wasser gewaschen; Das Waschwasser wird in einem Sedimentationsrohr gesammelt. Anschließend wird das „Bomben“-Rohr in das Sedimentationsrohr eingeführt und erneut heißes destilliertes Wasser zugegeben, so dass es das „Bomben“-Rohr vollständig bedeckt. Um die Reaktionsmischung vollständig aufzulösen, schütteln Sie sie. Nachdem sich die gesamte Mischung aufgelöst hat, wird das Reagenzglas mit einem dicken Platindraht angehoben und gründlich mit heißem destilliertem Wasser gewaschen. Dann wird das Reagenzglas mit einer Pinzette mit Platinspitzen an der Schlaufe genommen und innen mit heißem destilliertem Wasser gewaschen, das in die Originallösung gegossen wird.
BESTIMMUNG VON CHLOR. Die Lösung wird in einem Eisbad abgekühlt und mit konzentrierter Salpetersäure angesäuert. Die angesäuerte Lösung wird zur Fällung in ein anderes Reagenzglas filtriert; Zum Filtrat wird 1 ml 5 %ige Silbernitratlösung gegeben, im Wasserbad erhitzt und der Niederschlag filtriert.
Der Silberchloridniederschlag wird bis zur Gewichtskonstanz getrocknet und gewogen, anschließend wird der Chlorgehalt in der analysierten Substanz berechnet.
Bestimmung von Brom und Jod. Die Hauptlösung und das Waschwasser werden in Gegenwart von Phenolphthalein mit Salpetersäure neutralisiert, bis sie leicht rosa werden. 100 mg Hydrazinsulfat werden zugegeben und die Mischung 15 Minuten lang auf einem Dampfbad erhitzt. Nachdem diese Mischung zur Fällung in ein anderes Reagenzglas filtriert wurde, wird sie mit 0,5 ml konzentrierter Salpetersäure angesäuert, 1 ml einer 5%igen Silbernitratlösung hinzugefügt, alles erneut im Dampfbad erhitzt und der Niederschlag filtriert. getrocknet und gewogen (wie bei der Chlorbestimmung).
Titrimetrische Methoden.
Bestimmung von Chlor und Brom. Eine organische Substanz, die Chlor und Brom enthält, wird in den Reaktionskolben gegeben und bei 115–125 °C mit konzentrierter Schwefelsäure in Gegenwart einer Mischung aus Kaliumdichromat und Silberdichromat zersetzt. Die Verbrennung erfolgt im Sauerstoffstrom. Halogen (X) gelangt in eine Vorlage, die einen Überschuss an Natronlauge mit einer molaren Konzentration von 0,01 mol/l enthält, zu der 1 ml Wasserstoffperoxid gegeben wird:
2RX -> X 2 + X CO2+ X H 2 O (Oxidation),
X 2 + 2NaOH + H 2 O 2 = 2NaX + O 2 + 2H 2 O (Absorption).
Eine überschüssige Natronlauge wird mit einer Säurelösung gleicher Konzentration titriert und aus der verbrauchten Alkalimenge der Chlor- bzw. Bromanteil berechnet. Die Anwesenheit von Jod stört die Bestimmung nicht, da es zu Jodat oxidiert wird. Für flüchtige Stoffe oder Stoffe, die zusätzlich Stickstoff oder Schwefel enthalten, ist diese Methode nicht geeignet.
Bestimmung von Jod. Jodhaltige organische Stoffe werden in Gegenwart eines Katalysators in einer Sauerstoffatmosphäre verbrannt; Dabei entstehen Jod, Kohlenmonoxid (IV) und Wasser. Jod wird eingefangen und mit Brom zu Jodsäure HIO 3 oxidiert, diese wird mit Jodwasserstoff zu Jod reduziert, das jodometrisch bestimmt wird.
Der Prozess läuft nach folgenden Gleichungen ab:
2RI -> I 2 + X CO2+ X H 2 O (Verbrennung),
HIO 3 + 5HI = 3I 2 + 3H 2 O,
I 2 + 2Na 2 S 2 O 3 = 2NaI + Na 2 S 4 O 6 (iodometrische Titration).
§ 9. Analytische Bestimmung von Stickstoff
Qualitative Bestimmung von Stickstoff. In Verbrennungsprodukten stickstoffhaltiger organischer Stoffe liegt dieser in freiem Zustand vor. Um das Vorhandensein von Stickstoff in einer organischen Verbindung festzustellen, ist es daher notwendig, diese zu zerstören und den Stickstoff in eine Verbindung umzuwandeln, die durch einige qualitative Reaktionen leicht entdeckt werden kann.
Zum Beispiel, nach der Lassen-Methode Stickstoff wird auf diese Weise nachgewiesen: In einem an einem Ende verschlossenen Glasrohr wird ein wenig der untersuchten Substanz mit einem Stück Kaliummetall kalziniert; In diesem Fall verkohlt die Substanz und es entsteht Kaliumcyanid aus Kalium, Kohlenstoff und Stickstoff, die in der untersuchten organischen Substanz enthalten sind. Das Rohr wird in eine kleine Menge Wasser eingetaucht, wodurch es platzt und Kaliumcyanid sich im Wasser auflöst. Die Lösung wird filtriert, um Glassplitter und Kohle zu entfernen. Zu der resultierenden Lösung, die Kaliumcyanid enthält, werden Lösungen von Eisen(II)-Salzen, beispielsweise Eisen(II)-Sulfat, und Eisen(III)-Oxidsalzen, beispielsweise Eisen(III)-Chlorid, gegeben. Das in der Lösung enthaltene Kaliumhydroxid fällt Eisen(II)- und (III)-Hydroxide aus, und wenn Kaliumcyanid mit Eisen(II)-Salzen reagiert, entsteht Kaliumhexacyanoferrat(II) K4:
6KCN + FeSO 4 K 4 + K 2 SO 4.
Anschließend wird Salzsäure (Salzsäure) zugesetzt, wodurch wieder Eisensalze entstehen. Nach der Auflösung von Eisen(II)- und (III)-Hydroxiden bildete sich aus Kaliumhexacyanoferrat(II) und Eisen(III)-chlorid ein Niederschlag von Berliner Blau (Rest bleibt Eisen(III)-hexacyanoferrat Fe 4 3):
3K 4 + 4FeCl 3 = Fe 4 3 + 12KCl.
Die Bildung von Berliner Blau (ein wasserunlösliches Salz mit charakteristischer blauer Farbe) zeigt den Stickstoffgehalt der untersuchten Substanz an.
Quantitative Bestimmung von Stickstoff (Dumas-Methode). Mit Kupfer(II)-oxid vermischte organische Stoffe werden verbrannt, indem ein Kohlendioxidstrom durch ein Verbrennungsrohr geleitet wird; Verbrennungsprodukte werden in einem Gerät gesammelt, das mit einer konzentrierten Kaliumhydroxidlösung gefüllt ist. Kohlendioxid wird absorbiert, das Volumen des verbleibenden freien Stickstoffs gemessen und anschließend seine Masse berechnet.
Bei der Verbrennung stickstoffhaltiger organischer Stoffe wird am Ende des Verbrennungsrohrs eine reduzierte Kupferspirale angebracht: Entsteht eine geringe Menge Stickoxide, geben diese bei Kontakt mit dem heißen Kupfer Sauerstoff an dieses ab und es entsteht Stickstoff in einem freien Staat erhalten.
Der freigesetzte Stickstoff wird quantitativ in einem Stickstoffmessgerät (Messgefäß) über einer 50 %igen Kalilauge gesammelt. Die Methode wurde 1831 von J.B.A. Dumas entwickelt.
Die gasometrische Bestimmung von Stickstoff nach der Dumas-Methode ist auf organische Verbindungen anwendbar, die Stickstoff in jeglicher Form enthalten, nämlich: Amino-, Nitroso-, Azo-, Cyanoverbindungen, Alkylnitrite sowie Nitrate und heterozyklische stickstoffhaltige Verbindungen.
Bei der Bestimmung von Stickstoff in organischen Verbindungen (hauptsächlich Aminen) nach der Kjeldahl-Methode wird der Analyt mit konzentrierter H 2 SO 4 in Gegenwart eines Katalysators, meist Quecksilber oder dessen Salze, zersetzt. Dabei wird nach der Alkalisierung Ammoniak isoliert, abdestilliert, mit einer Lösung von H 2 SO 4 oder HCl absorbiert und titrimetrisch oder kolorimetrisch mit Nesslers Reagenz – einer alkalischen Lösung von Kaliumtetraiodomercurat(II)-dihydrat K 2 [HgI 4 ] 2H 2 O. Um genauere Ergebnisse zu erhalten, wird die stickstoffhaltige Substanz manchmal mit einem Reduktionsmittel, beispielsweise HI, vorbehandelt. Die Methode wurde 1883 von I. Kjeldahl entwickelt.
Es gibt halbautomatische Analysegeräte, mit denen Sie den Stickstoffgehalt gleichzeitig mit dem Kohlenstoff- und Wasserstoffgehalt bestimmen können.
§ 10. Analytische Bestimmung von Sauerstoff
Eine der ersten Methoden zur Bestimmung von Sauerstoff in organischen Verbindungen ist die Reduktion in einem Wasserstoffstrom (Hydrierung) bei einer Temperatur von 1120 °C. Dabei entsteht Wasser.
Darüber hinaus können organische Verbindungen durch Fusion mit Kalium- oder Natriummetall zerstört werden. In diesem Fall bildet Sauerstoff ein Metalloxid, das leicht nachweisbar ist. Enthält die Verbindung jedoch gleichzeitig Sauerstoff und Stickstoff, kann ein Alkalimetallcyanat, das Salz der Cyansäure HNCO, entstehen.
Nach der Unterzaucher-Methode wird eine sauerstoffhaltige organische Verbindung in einer Stickstoffatmosphäre einer Pyrolyse unterzogen. Gasförmige Produkte werden über auf 1110 – 1150 °C erhitzte Kohle geleitet. Dabei entsteht Kohlenoxid (II), das dann mit festem Jodoxid (V) (Jodsäureanhydrid) I 2 O 5 reagiert:
5CO + I 2 O 5 = 5CO 2 + I 2.
Das freigesetzte Jod wird titrimetrisch mit Natriumthiosulfat Na 2 S 2 O 3 bestimmt:
2Na 2 S 2 O 3 + I 2 = 2NaI + Na 2 S 4 O 6.
Diese Methode wurde 1940 von J. Unterzaucher entwickelt.
Einige Forscher bevorzugen die Verwendung eines vertikalen T-förmigen Quarzrohrs, das mit Kohlenstoff gefüllt ist (Abbildung 6).
Direkte Mikrobestimmung von Sauerstoff in organischen Verbindungen.
Der Stoff wird einer reduktiven Zersetzung über Kohle in einer inerten Atmosphäre unterzogen. In diesem Fall wird der gesamte Sauerstoff in Kohlenmonoxid (II) umgewandelt:

![]()
M.O. Korshun leistete einen großen Beitrag zur Entwicklung der Methode zur Mikrobestimmung von Sauerstoff und anderen Elementen. Sie schlug die gravimetrische Bestimmung von Sauerstoff auf der Grundlage von drei Faktoren vor:
1) durch den Massenverlust von I 2 O 5 (siehe vorherige Gleichung);
2) durch Gewichtszunahme von Kupfer:
Cu + I = CuI 2;
3) durch Ascarit-Gewichtszunahme*:
CO 2 + 2NaOH = Na 2 CO 3 + H 2 O.
§ 11. Analytische Bestimmung von Schwefel
Schwefelhaltige organische Stoffe werden nach der Carius-Methode oxidiert „Nassbrennen“ in konzentrierter Salpetersäure in einem verschlossenen dickwandigen Glasrohr in Gegenwart von Bariumchlorid bei einer Temperatur von 280–300 °C. Der in der Substanz enthaltene Schwefel wird zu Schwefelsäure oxidiert, die dann mit Bariumchlorid zu Bariumsulfat reagiert:
H 2 SO 4 + BaCl 2 = BaSO 4 + 2HCl.
Nach dem Abkühlen wird das Röhrchen geöffnet, der Niederschlag entnommen, gewaschen, getrocknet und gewogen (gravimetrische Analyse).
Von Methode „Trockenbrennen“. Eine organische Substanz, die Schwefel, aber keine Halogene und Stickstoff enthält, wird in einer Sauerstoffatmosphäre in Gegenwart eines Platinkatalysators oxidiert. Dabei wird Schwefel in gasförmige Oxide umgewandelt, die von einer wässrigen Wasserstoffperoxidlösung absorbiert werden:
SO 2 + SO 3 + H 2 O 2 + H 2 O = 2H 2 SO 4.
Das Endprodukt der Schwefeloxidation ist somit Schwefelsäure, die mit einer Natriumhydroxidlösung mit einer molaren Konzentration von 0,01 mol/l titriert wird (titrimetrische Analyse):
H 2 SO 4 + 2NaOH = Na 2 SO 4 + 2H 2 O.
§ 12. Analytische Bestimmung von Phosphor und Arsen
Zur Bestimmung von Phosphor wird organisches Material entweder durch Fusion mit festen oxidierenden Gemischen oder durch Behandlung mit einem Gemisch aus Wasserstoffperoxid und konzentrierter Schwefelsäure zersetzt. Dabei wird der in der organischen Substanz enthaltene Phosphor in Orthophosphorsäure umgewandelt, die dann gewichtsmäßig (gravimetrische Analyse) in Form von zuvor mit Ether getrocknetem Ammoniumphosphoromolybdat bestimmt wird.
Zur Bestimmung von Arsen werden organische Stoffe ähnlich wie phosphorhaltige Verbindungen oxidiert. In diesem Fall verwenden sie entweder „Nassverbrennung“ (Carius-Methode) oder Fusion in einer „Bombe“. Die resultierende Arsensäure H 3 AsO 4 wird gravimetrisch (nach Gewicht) in Form des Magnesiumsalzes der Pyromesinsäure bestimmt:
AsO 3- 4 + Mg 2+ + NH 4 + = MgNH 4 AsO 4,
2MgNH 4 AsO 4 Mg 2 As 2 O 7 + H 2 O + 2NH 3 ;
oder jodometrisch (titrimetrische oder volumetrische Methode):
H 3 AsO 4 + 2HI = H 3 AsO 3 + I 2 + H 2 O,
I 2 + 2Na 2 S 2 O 3 = 2NaI + Na 2 S 4 O 6.
§ 13. Bestimmung der Struktur organischer Stoffe
Chemie ist ein Bereich wissenschaftlicher Erkenntnisse und praktischer Tätigkeit, der sich durch die Synthese neuer Verbindungen und die Schaffung darauf basierender Materialien entwickelt. Die Hauptaufgabe eines Forschungschemikers nach der Synthese einer Verbindung besteht darin, die Struktur des Moleküls aufzuklären.
Die Bestimmung der vollständigen Struktur eines Moleküls, einschließlich der geometrischen und elektronischen Struktur, ist eine komplexe Aufgabe, die nicht vollständig gelöst werden kann. Daher können die Details der Molekülstruktur einer mehr oder weniger komplexen Verbindung endlos untersucht werden, wodurch immer mehr neue Informationen hinzugefügt werden.
Betrachten wir die physikalischen Grundlagen der wichtigsten Methoden zur Bestimmung der Struktur von Molekülen organischer Verbindungen. Diese Methoden werden physikalische Forschungsmethoden genannt. Auch chemische Methoden zum Nachweis der geometrischen Struktur eines Moleküls (geometrische Struktur) haben nicht an Bedeutung verloren.
Zum Zeitpunkt der Entstehung der Strukturtheorie von A. M. Butlerov wurde die wichtige Wahrheit erkannt, dass die Struktur eines Moleküls alle seine chemischen Eigenschaften bestimmt und aus der Summe der chemischen Eigenschaften eine korrekte Schlussfolgerung über die Struktur eines Moleküls gezogen werden kann Substanz. Diese Wahrheit hat im Zeitalter der physikalischen Forschungsmethoden keineswegs ihre Bedeutung verloren. Analyse und Synthese bleiben in der Chemie konstant; Diese Konzepte bilden, wie wir wissen, die Grundlage des Denkprozesses, die Grundlage allen Wissens. Bei der Synthese immer komplexerer Stoffe kennt der Chemiker im Voraus die Ausgangsfragmente, aus denen die Struktur der neuen Verbindung entsteht. Daher nimmt es eine Struktur im Voraus an. Nach der Synthese bleibt nur noch der Beweis. Wenn die Struktur einer natürlichen Verbindung ermittelt wird, über die keine Informationen vorliegen, werden zunächst deren chemische Eigenschaften untersucht, die Rückschlüsse auf das Vorhandensein bestimmter Strukturelemente zulassen. Der wichtigste Schritt hierbei ist die chemische Analyse – die Zerlegung eines komplexen Moleküls in Fragmente, in einzelne „Bausteine“ und die Feststellung der Art ihrer Kombination im Molekül.
In den letzten Jahrzehnten wurde die chemische Analytik ergänzt Massenspektralanalyse– eine physikalische Methode, die auf der Messung der Masse geladener Materieteilchen basiert, die entstehen, wenn komplexe Moleküle von einem starken Elektronenfluss bombardiert werden. Traditionell wird diese Methode immer zusammen mit Methoden der optischen Spektroskopie betrachtet. Die oben genannten Methoden ermöglichen den Nachweis des Vorhandenseins einzelner wesentlicher Elemente der geometrischen (IR-Spektroskopie, Raman-Spektroskopie) und elektronischen (Elektronenspektroskopie, Kernspinresonanzspektroskopie, Elektronenparamagnetische Resonanzspektroskopie, Röntgenelektronenspektroskopie usw.) Struktur des Moleküls und um ein besseres Verständnis davon zu erlangen.
Kernspinresonanzspektroskopie (NMR-Spektroskopie)– eine der modernsten Methoden zur Untersuchung der Struktur organischer Verbindungen. Neben der Elektronen- und Schwingungsspektroskopie ermöglicht die mit der Resonanzspektroskopie verwandte NMR-Methode die Lösung subtilster Fragen der Strukturchemie, insbesondere Fragen zum elektronischen Zustand von Atomen in einem Molekül. Die Methode eignet sich zur Untersuchung von Molekülen, die Atome mit einer ungeraden Anzahl von Protonen oder Neutronen enthalten. Die Kerne solcher Atome haben ein magnetisches Moment und sind paramagnetisch.
Die theoretischen Grundlagen dieser Methoden zur Untersuchung der Struktur organischer Verbindungen sind komplex und werden in einschlägigen Hochschulstudiengängen diskutiert.
* Ascarit ist Asbest, der mit geschmolzener NaOH imprägniert ist. – Notiz Hrsg.
Die Grundlagen der Definition wurden von F. Pregl entwickelt. Eine Probe von 3-5 mg der Substanz wird bei einer Temperatur von 900 °C in einem aus Wasserstoff, Wasser und Kohlendioxid gereinigten Sauerstoffstrom verbrannt. Wasserstoff wird von Sauerstoff gereinigt, indem das Gas bei 800 °C über einen Platinkatalysator geleitet wird. Die vollständige Entfernung von Kohlendioxid und Wasser erfolgt durch Durchleiten durch wasserfreies Magnesiumperchlorat ( Anhydron) und durch mit geschmolzener Natronlauge imprägniertes Asbest ( Ascarit).
Nach dem Verbrennungsrohr sind Rohre mit Absorbern installiert: Anhydron und Ascarit. Die Gewichtszunahme des ersten Absorbers entspricht der Wassermenge, aus der der Wasserstoffgehalt in einer Probe des Stoffes berechnet wird; Die Gewichtszunahme des zweiten Absorbers ergibt die Kohlendioxidmenge, die zur Bestimmung des Kohlenstoffgehalts im analysierten Stoff verwendet wird.
Halogene und Schwefel können durch Zersetzung der Substanz nach Carius bestimmt werden. Halogene in Form von Silberhalogenid – durch Gravitation oder durch Titrieren von überschüssigem Silbernitrat. Schwefel wird in Form von Bariumsulfat bestimmt. Nach der Verbrennung der Probe werden Gase über eine Schicht aus Kupfermetall geleitet, wo Stickoxide zu freiem Stickstoff reduziert werden. Stickstoff wird nach der volumetrischen Methode anhand der Menge an nicht absorbiertem Gas bestimmt.
Bestimmung des Molekulargewichts
Um das Molekulargewicht einer Verbindung zu bestimmen, werden häufig Kryoskopiemethoden verwendet, die auf dem Raoult-Gesetz basieren. Bestimmen Sie dazu den Gefrierpunkt des Lösungsmittels und anschließend der Lösung. Die Differenz ist direkt proportional zur Anzahl der Moleküle der Substanz, die in einer bestimmten Lösungsmittelmasse gelöst sind. Die Molekularmasse wird durch die Formel bestimmt:
Wo R- Gewicht des Stoffes; P – Lösungsmittelgewicht; K – kryoskopische Konstante;
In ähnlicher Weise wird bei der ebullioskopischen Methode das Molekulargewicht durch die Differenz zwischen den Siedepunkten eines reinen Lösungsmittels und einer Lösung bestimmt.
Für hochmolekulare Verbindungen sind die oben beschriebenen Methoden überhaupt nicht anwendbar. Dabei kommen drei Methoden zum Einsatz: viskometrische, osmotische und Sedimentation, die wiederum nicht auf Stoffe mit normalem Molekulargewicht anwendbar sind.
Derzeit wird Massenspektrometrie am häufigsten zur Bestimmung der Molekülmasse einer unbekannten Substanz eingesetzt.
Methoden zur Isolierung einzelner Substanzen
Um Verbindungen zu isolieren, werden folgende physikalische Methoden verwendet: verschiedene Arten der Destillation – fraktionierte Destillation bei Atmosphärendruck, im Vakuum, im Hochvakuum, Molekulardestillation, Kristallisation, Extraktion, Chromatographie. Darüber hinaus gibt es viele spezielle Methoden, die die Besonderheiten der Funktionsgruppe berücksichtigen.
· Molekulare Destillation . Für Stoffe, die sich auch im Hochvakuum am Siedepunkt zersetzen, wird die „Molekulardestillation“ eingesetzt. Sein Prinzip besteht darin, dass unter starkem Vakuum (10 -5 -10 -8 mm Hg) von der erhitzten Oberfläche der zu destillierenden geschmolzenen Substanz die Moleküle bei einer Temperatur, die viel niedriger ist als der Siedepunkt der jeweiligen Verbindung, in die Gasphase übergehen . Der Dampf des Stoffes kondensiert dann an der kalten Oberfläche. Dadurch ist es möglich, Substanzen mit relativ großem Molekulargewicht und fragiler Struktur zu reinigen.
· Wasserdampfdestillation . Bekanntlich siedet ein Stoff bei einer Temperatur, bei der sein Dampfdruck dem Atmosphärendruck entspricht. Wenn Sie zwei nicht mischbare Flüssigkeiten erhitzen, sieden sie bei einer Temperatur, bei der der Gesamtdampfdruck beider Flüssigkeiten dem Atmosphärendruck entspricht. Als zweite Flüssigkeit wird Wasser verwendet. Somit kann die Destillation dieses Flüssigkeitsgemisches unterhalb von 100 °C durchgeführt werden. Die Menge beider Stoffe im Destillat wird durch das Verhältnis des Produkts aus dem Dampfdruck jedes Stoffes und seinem Molekulargewicht bestimmt.
· Kristallisation . Umkristallisation wird zur Reinigung von Feststoffen eingesetzt. Die Methode basiert auf der Tatsache, dass bei den meisten Verbindungen die Löslichkeit der Substanz abnimmt, wenn ihre Lösungen abgekühlt werden.
· Extraktion . Eine Trennmethode, die auf dem Unterschied der Verteilungskoeffizienten einer Substanz zwischen zwei nicht mischbaren Flüssigkeiten basiert.
Chromatographie
Ø Chromatographie – Trennmethode basierend auf unterschiedlichen Bewegungsgeschwindigkeiten der Konzentrationszonen der Komponenten der untersuchten Mischung im Fluss der mobilen Phase ( Eluent) relativ zur stationären Phase.
§ Durch zu lösende Aufgaben zuordnen präparativ(quantitative Stofftrennung) und analytisch Chromatographie (Nachweis von Stoffen sowie quantitative und qualitative Eigenschaften von Gemischen).
§ Nach den Grundsätzen der Trennung Chromatographie ist unterteilt in Adsorption, Verteilung, Ionenaustausch Und Sieb.
v Adsorptionschromatographie . Die zu trennenden Stoffe müssen sich in ihrer Affinität zum festen Adsorbens, der stationären Phase, unterscheiden. Als Adsorptionsmittel werden üblicherweise Aluminiumoxid und Kieselgel verwendet. Deutlich seltener kommen Aktivkohle, Bariumsulfat, Magnesiumsilikat und Polyamide zum Einsatz.
Die Adsorptionsfähigkeit von Stoffen an einem polaren Adsorbens wird maßgeblich von ihrer Polarität bestimmt. Aufgrund ihrer Adsorptionsfähigkeit können Stoffe mit unterschiedlichen funktionellen Gruppen in folgender Reihenfolge angeordnet werden:
RH< ROCH 3 < R-NO 2 < R-N(CH 3) 2 < R-COOCH 3 < R-NH 2 < R-OH < R-CONH 2 < R-COOH.
Hinsichtlich der Polarität und damit der Elutionsfähigkeit bilden Eluentenlösungsmittel die folgende Reihe:
H 2 O > CH 3 OH > C 2 H 5 OH > CH 3 COCH 3 > CH 3 COOC 2 H 5 > C 2 H 5 OC 2 H 5 > CHCl 3 > CCl 4 > Cyclohexan > N-Hexan
Die Elution erfolgt entweder mit einem Eluenten (einer Mischung von Eluenten) oder nacheinander mit mehreren Eluenten, wobei von weniger polar zu polarer übergegangen wird, oder mit einer Mischung aus zwei Lösungsmitteln (wobei die Konzentration des polareren Lösungsmittels sukzessive erhöht wird).
· Hauptvarianten der Adsorptionschromatographie .
§ Säulenadsorptionschromatographie. Das Adsorptionsmittel wird in eine Säule gegeben. Zuerst wird der zu trennende Stoff darauf aufgetragen, dann wird der Eluent durchgeleitet, der sich unter dem Einfluss der Schwerkraft bewegt oder von einer speziellen Pumpe unter Druck gepumpt wird.
Die Stofftrennung wird entweder durch physikalisch-chemische Methoden (UV-Detektion, Refraktometrie) oder durch analytisch-chromatographische Methoden überwacht.
§ Dünnschichtchromatographie (TLC). Das Sorptionsmittel wird in einer dünnen Schicht auf ein Glas-, Aluminium- oder Kunststoffsubstrat aufgetragen.
Die Sorptionsschicht kann lose sein oder mit speziellen Chemikalien (Stärke, Gips) fixiert werden. Eine Probe der Substanz wird auf den Boden der Platte aufgetragen und diese dann in eine Box mit einem Elutionsmittel gegeben. Das Lösungsmittel steigt aufgrund der Kapillarkräfte entlang der Platte ( aufsteigend Chromatographie), wodurch eine Trennung entsteht. Bei schwer trennbaren Stoffen greifen Sie zu zweidimensional Chromatographie, wenn die Substanz zunächst in einer Richtung eluiert wird und dann die Elution in einer senkrechten Richtung durchgeführt wird.
Unter modernen Bedingungen werden üblicherweise industriell hergestellte Platten für die präparative oder analytische DC verwendet.
· Identifizierung und Charakterisierung von Stoffen. Farbige Verbindungen werden direkt während der Chromatographie beobachtet. Farblose Stoffe müssen „identifiziert“ – in farbige Verbindungen umgewandelt werden.
Abhängig vom Sorbens, Fixiermittel und der Art der abgetrennten Substanz werden verschiedene Methoden zum „Nachweis“ verwendet. Beispielsweise werden Kohlenhydrate bei hohen Temperaturen verkohlt, auch nach dem Besprühen mit Schwefelsäurelösungen. Aminosäuren ergeben nach der Behandlung mit a gefärbte Produkte Ninhydrinlösung. Anhand der Farbintensität der mit bestimmten Reagenzien getrennten Verbindungen wird deren Gehalt in der Mischung beurteilt.
Zur Charakterisierung von Stoffen wird der Begriff „ chromatographische Mobilität", das als Rf bezeichnet wird, ist das Verhältnis der Reichweite der Substanzzone zur Reichweite des Eluenten.
v Verteilungschromatographie . Diese Version der Chromatographie basiert auf der Verteilung von Substanzen zwischen der mobilen Phase (Gas, Flüssigkeit) und der stationären Phase – einer Flüssigkeit, die auf einem festen inerten Träger gehalten wird. Am weitesten verbreitet sind die Verteilungschromatographie auf Papier und die Gas-Flüssigkeits-Chromatographie.
§ Papierchromatographie. Grundlage der Papierchromatographie ist die Verteilung eines Gemisches getrennter Stoffe zwischen auf Papier adsorbiertem Wasser und einem mit Wasser gesättigten Lösungsmittel. Mit dieser Methode gelang die Trennung und Identifizierung von Aminosäuren und Monosacchariden erfolgreich. Derzeit hat diese Option der Chromatographie ihre Relevanz verloren.
§ Gas-Flüssigkeits-Chromatographie. Hierbei handelt es sich um eine Verteilungschromatographie zwischen einer stationären flüssigen Phase auf einem Träger und einem Gas (normalerweise Helium, Stickstoff oder Wasserstoff).
Zur Charakterisierung der abgetrennten Stoffe verwenden Sie „ Aufbewahrungszeit" Dabei handelt es sich um die Zeit vom Einleiten des Gemischs in die Säule bis zum Verlassen der Säule und dem Durchlaufen der Substanz durch einen geeigneten Detektor, beispielsweise einen Detektor, der Änderungen der Wärmeleitfähigkeit aufzeichnet. Diese Option ist eine der am weitesten verbreiteten chromatographischen Methoden, insbesondere für analytische Zwecke.
v Ionenaustauschchromatographie . Die Methode basiert auf der Verteilung geladener Substanzen (Ionen) zwischen der mobilen und der stationären Phase in Abhängigkeit von ihrer Affinität zu den ionischen Zentren der stationären Phase.
Je nach Art der Ionenaustauscher gibt es solche kationisch Und anionisch Chromatographie. Als Elutionsmittel werden häufig Wasser, Lösungen von Säuren und Laugen sowie Pufferlösungen verwendet. Die gebräuchlichsten Ionenaustauschermaterialien sind Kationenaustauscher und Anionenaustauscher auf Basis vernetzter Polymere mit ionogenen funktionellen Gruppen sowie modifizierter Cellulose.
v Siebchromatographie (Gelchromatographie) . Eine Besonderheit der Gelchromatographie besteht darin, dass in Gelen, die aus dreidimensional „vernetzten“ Makromolekülen bestehen, Poren bestimmter Größe vorhanden sind, in die die kleineren Moleküle der getrennten Moleküle eindringen und die größeren nicht. Daher passieren bei der Gelchromatographie im Gegensatz zur Adsorptionschromatographie die größeren Moleküle zuerst und die kleinen zuletzt die Säule. Die Trennsäule ist mit Körnern aus lyophilem oder hydrophilem Gel gefüllt. Beispiele für solche chromatographischen Materialien sind modifizierte natürliche Geliermittel – Agar, Dextrine, Sephadexe (vernetzte Dextrane) und synthetische Siebe auf Basis von Polyacrylamid oder „vernetztem“ Polystyrol.
Berzelius J. (1779 1884) – schwedischer Chemiker. Die wissenschaftliche Forschung deckt alle Hauptprobleme der Chemie der ersten Hälfte des 19. Jahrhunderts ab.
Wehler F. (1800–1882) – deutscher Chemiker. Arbeitet in der anorganischen und organischen Chemie. Zusammen mit J. Liebig begründete er die Isomerie der Salze explosiver Säuren.
3 Gmelin L. (1788-1853) – deutscher Chemiker. Er veröffentlichte Nachschlagewerke zu experimentellen Daten, die mehrere Auflagen erlebten.
Liebig J. (1803–1873) – deutscher Chemiker. Schöpfer der Radikaltheorie, Begründer der Agrochemie. Untersuchte organische Säuren.
Butlerov A. (1828-1886) – russischer Chemiker, Schöpfer der Theorie der Struktur organischer Verbindungen. Vorhergesagte Isomerie vieler Verbindungen.
Gerard S. (1816–1856) – französischer Chemiker. Arbeitete für Yu. Liebig, hörte Vorlesungen von J. Dumas. Erstellte eine Typentheorie. Viele russische Chemiker studierten bei C. Gerard.
Schorlemmer K. (1834 – 1892) – deutscher organischer Chemiker. Er arbeitete auf dem Gebiet der Alkane und hat Arbeiten zur Geschichte der Chemie.
Lossen V. (1838 – 1905) – deutscher Chemiker. Die Hauptarbeiten beziehen sich auf die Erforschung von Alkaloiden und entdeckten die Umlagerung von Hydroxamsäuren.
Carius L. (1829–1875) – deutscher Chemiker. Entwickelte eine Methode zur Bestimmung von Schwefel, Halogenen und anderen Elementen in organischen Verbindungen (1860)
Beilstein F. (1838–1906) – russischer organischer Chemiker. Hauptwerke auf dem Gebiet der Synthese aromatischer Verbindungen. Initiator und erster Verfasser des mehrbändigen Handbuchs der organischen Chemie, bekannt als Belstein Handbook.
Pregl F. (1869-1930) – deutscher Chemiker. Begründer der Mikroanalyse organischer Verbindungen. Nobelpreis 1923
Raoul F. (1830–1901) – französischer Chemiker. Die Hauptrichtung der Forschung ist das Studium von Lösungen.
ANALYSE ORGANISCHER STOFFE
(veraltete Organisationsanalyse), Qualitäten. und Mengen. Festlegung der Zusammensetzung der Org. in-in und Etablierung ihrer Struktur.
Bei der Bestimmung von Qualitäten. Zusammensetzung der Org. Sie verwenden eine Vielzahl von Methoden, die auf der Chemie basieren. p-tionen, begleitet von der Bildung von Produkten mit charakteristischen Eigenschaften (Farbe, Geruch, Schmelztemperaturen usw.), und auf physikalischen Messungen. und physikalisch-chemisch (chromatographische, spektrale usw.) Eigenschaften der identifizierten Verbindungen.
Mit Mengen, Analyse von org. Es wird die Menge des Reagenzes ermittelt, die mit der ermittelten Org in die Verteilung gelangt. Anschl. oder Maßdifferenz. körperlich und physikalisch-chemisch Merkmale, die mit der Anzahl der zu bestimmenden Verbindungen verbunden sind.
O.v. A. beinhaltet elementare Analyse, Strukturgruppe (einschließlich funktioneller und stereospezifischer), Molekularanalyse, Phasenanalyse Und Strukturanalyse.
Historisch gesehen wurden als erste Methoden zur Elementaranalyse von organischen Stoffen entwickelt. in-in (A. Lavoisier, Ende des 18. Jahrhunderts), basierend auf ihrer Oxidation und gravimetrischen, titrimetrischen. oder gasometrisch Bestimmung der gebildeten einfachen Verbindungen. einzelne Elemente. Erste elementare Methoden mikrochemische Analyse(Mikroanalyse) wurde ursprünglich von F. Pregl entwickelt. 20. Jahrhundert Ab der 2. Hälfte. 20. Jahrhundert Für die Elementaranalyse werden häufig automatische Methoden eingesetzt. Analysegeräte basierend auf der Verbrennung der analysierten Probenorg. in-va und gaschromatographisch. Trennung und Bestimmung von Verbrennungsprodukten. Der Analysator ist mit einem Computer und einer Automatik ausgestattet Probeneinführungssystem.
Isotopenanalyse von org. Der Zweck besteht darin, den Gehalt einzelner Isotope in ihnen sowie das Verhältnis derselben Org zu bestimmen. Verbindungen, die verschiedene oder Kombinationen davon enthalten. Zu diesem Zweck werden am häufigsten Massenspektrometrie oder multiple Gas-Flüssigkeits-Chromatographie verwendet (z. B. bei der Trennung gewöhnlicher und deuterierter Formen von Methan oder Benzol). Naib. Chromatographie-Massenspektrometrie ist effektiv.
Die meisten Methoden der Funktionsanalyse basieren auf Interaktion. einzelne Funktionen Gruppenorg. Anschl. mit geeigneten Reagenzien. Solche Bezirke können selektiv oder begrenzt selektiv, also charakteristisch, sein. nur für einen oder mehrere. funktionell Gruppen.
Am häufigsten werden Lösungen verwendet, die mit der Bildung oder dem Verschwinden von Substanzen, Basen, Oxidationsmitteln, Reduktionsmitteln, Wasser, Gasen und seltener Sedimenten und farbigen Substanzen verbunden sind. Die resultierenden Verbindungen und Basen bestimmen Säure-Base-Titration in aquatischen oder nichtwässrigen Umgebungen. In einer nichtwässrigen Umgebung trennen Sie potentiometrische Messungen Titration von Verbindungen und Basen unterschiedlicher Stärke, wenn sie zusammen vorliegen.
Im Fall der Oxidations-Reduktion. Bei Lösungen, deren Geschwindigkeit gering ist, wird meist eine Rücktitration eingesetzt, d. h. der Überschuss des Reagens wird titriert. Zur Entstehung bzw. Aufnahme von Wasser in den Bezirken der Org. Anschl. basierend auf der Definition des Plurals. funktionell Gruppen verwenden Fischers Reagenz(siehe auch Aquametrie).
Methoden, die auf Strömungen basieren, die mit der Freisetzung oder Absorption von Gas einhergehen, werden selten verwendet, da die Messung von Volumen oder Druck meist sperrige Geräte erfordert.
Gravimetrische Messungen basieren auf der Bildung von Sedimenten. Methoden zur Bestimmung einer kleinen Anzahl von Funktionen. Gruppen. Die in diesen Fällen eingesetzten schwerlöslichen Verbindungen sind in der Regel metallischer Natur. Kohlen- und Sulfonsäuren, Salze bio. Grundlagen, komplexe Zusammenhänge. (einschließlich Chelatverbindungen).
Bildung farbiger Verbindungen. oft recht spezifisch und ermöglicht eine gezielte Bestimmung der Funktion. photometrische Gruppen Methoden. Weit verbreitet (insbesondere in der Mikroanalytik) sind Lösungen, die zur Bildung fluoreszierender Verbindungen führen, da die Empfindlichkeit der Funktionsbestimmung eingeschränkt ist. Die Gruppe ist in diesem Fall ziemlich groß.
Eine besondere Art von Funktionalität. Analyse berücksichtigen Methoden auf der Grundlage vorläufiger. Wechselwirkung der zu bestimmenden Substanz mit Reagenzien und Bestimmung des resultierenden Produkts. Zum Beispiel aromatisch nach der Nitrierung kann polarographisch bestimmt werden, und die Beziehung zwischen der Aminogruppe und Naphthalinsulfonylchlorid kann fluormetrisch bestimmt werden.
Nachfolgend finden Sie Beispiele für die meisten. häufig verwendete Funktionsmethoden. Analyse.
Die Bestimmung von aktivem Wasserstoff in Alkoholen, Aminen, Amiden, Kohlen- und Sulfonverbindungen, Mercaptanen und Sulfonamiden basiert auf deren Wechselwirkungen. mit Grignard-Reagenzien (meist Methylmagnesiumiodid; s Cerevitin-Methode)oder mit LiAlH 4 und Messung des freigesetzten Methan- bzw. Wasserstoffvolumens. Aktiv in Acetylen und seinen Homologen wird durch die Lösung mit den Salzen Ag(I), Hg(I) oder Cu(I) mit letzterem titrimetrisch bestimmt. Bestimmung der Getrennten.
Verbindungen mit ungesättigten Kohlenstoff-Kohlenstoff-Bindungen sind am häufigsten bromiert, jodiert oder hydriert. In den ersten beiden Fällen wird nicht umgesetztes Br 2 bzw. I 2 iodometrisch bestimmt und während der Hydrierung das Volumen an absorbiertem H 2 gemessen. Die Anzahl der Doppelbindungen lässt sich durch Zugabe von Quecksilbersalzen zu letzteren bestimmen. Titration der freigesetzten Substanz.
Bei der Bestimmung von Hydroxylgruppen verwenden sie am häufigsten Essigsäure-, Phthalsäure- oder Pyromellitsäureanhydrid, deren Überschuss titriert wird. Sie können Säurechloride verwenden. Hydroxygruppen in Phenolen werden üblicherweise mit Basenlösungen in einem nichtwässrigen Medium titriert. Phenole lassen sich leicht bromieren und mit Diazoniumsalzen kombinieren, daher werden sie mit Lösungen von Br 2 oder Diazoniumsalzen titriert oder der untersuchten Lösung wird ein Bromid-Bromat-Gemisch zugesetzt, der Überschuss wird iodometrisch bestimmt (siehe auch Falins Reaktion).
Kohlenhydrate können durch Oxidation mit Natriumperiodat und anschließender Oxidation bestimmt werden. Titration von überschüssigem Oxidationsmittel oder gebildeten Verbindungen. Zahlreiche wurden entwickelt. Variationen dieser Methode (siehe zum Beispiel Malaprada-Reaktion).
Zur Ermittlung der Org. Peroxyverbindungen (einschließlich Peroxysäuren) nutzen am häufigsten ihre Wechselwirkung. mit KI und anschließend Titration des freigesetzten I 2 mit Na 2 S 2 O 3 -Lösung.
Die Analyse von Alkoxyverbindungen besteht aus Wechselwirkungen. der analysierten Substanz mit Iodwasserstoffsäure zu Alkyliodiden (vgl. Zeisel-Methode). Letztere werden mit unterschiedlichen Methoden bestimmt – gravimetrisch (in Form von AgI) oder titrimetrisch (Säure-Base-Titration). Kohlenstoffverbindungen können auf ähnliche Weise bestimmt werden. Zur Identifizierung von C 1 -C 4 -Alkoxygruppen werden die resultierenden Alkyliodide in quartäre Ammoniumverbindungen umgewandelt, die mittels Dünnschicht- oder Papierchromatographie analysiert werden.
Die Definition von Epoxidgruppen basiert auf ihrer Reaktion mit Chlorwasserstoff unter Bildung von Chlorhydrinen; Nach Beendigung der Lösung wird die überschüssige HCl mit Alkalilösung titriert.
Zur Bestimmung von Carbonylverbindungen. (Aldehyde und Ketone) max. Häufig kommt die Oximation zum Einsatz, also deren Umwandlung in Wechselwirkung. mit Hydroxylaminhydrochlorid; Das bei der Reaktion freigesetzte HCl wird mit einer Alkalilösung titriert (der Endpunkt der Titration wird über einen Indikator oder potentiometrisch eingestellt). Es gibt eine Vielzahl von Modifikationen dieser Methode. Aldehyde können auch durch die Lösung mit Na-Bisulfit und anschließend bestimmt werden. Säure-Base-Titration. Weniger häufig verwendet werden Aldehyde mit Ag+-Ionen, Reaktion mit Hydrazinen und die Bildung von Schiffschen Basen.
Chinone werden mit Ti(III)-Chlorid oder V(II)-Sulfat reduziert; der Überschuss des Reduktionsmittels wird titrimetrisch bestimmt. Chinone können auch jodometrisch bestimmt werden.
Zur Bestimmung von Kohlenstoffverbindungen und deren Salzen werden max. Die Säure-Base-Titration wird häufig in nichtwässrigen Medien eingesetzt.
Für die Analyse von Kohlenstoffderivaten wurde eine Vielzahl von Methoden entwickelt. Anhydride werden nach ihrer Hydrolyse zu einer Lösung mit Alkalilösungen titriert. Bei der Analyse eines Gemisches aus einer Säure und ihrem Anhydrid wird die Summe beider Stoffe durch Säure-Base-Titration bestimmt, anschließend das Anhydrid mit Morpholin oder Anilin vermischt und die freigesetzten Säuren titriert. Im letzteren Fall können Sie den Basenüberschuss auch durch Titration mit HCl-Lösung bestimmen. Auf die gleiche Weise werden Säurehalogenide oder deren Mischungen mit Verbindungen bestimmt. In diesem Fall werden anstelle von Lösungen mit Aminen häufig Wechselwirkungen eingesetzt. Säurehalogenid mit Alkohol und zuletzt. separate Titration von freiem Kohlensäure und die freigesetzte halogenreiche Säure mit Alkalilösung.
Die Bestimmung von Kohlenstoffestern basiert auf deren Hydrolyse mit einer Alkalilösung, der Überschuss wird mit einer Lösung titriert. Geringe Mengen an Estern werden üblicherweise spektrophotometrisch in Form von Fe(III)-Salzen von Hydroxamsäuren bestimmt, die bei der Wechselwirkung entstehen. Ester mit Hydroxylamin.
Zur Bestimmung stickstoffhaltiger org. Es wurde eine große Anzahl von Methoden vorgeschlagen. Reduktionsfähige Verbindungen (Nitro-, Nitroso-, ) werden titan- oder vanadatometrisch bestimmt: Eine überschüssige Lösung von Ti(III)- oder V(II)-Salz wird zugegeben und das nicht umgesetzte Reduktionsmittel wird mit einer Lösung von Fe(III) titriert. Salz.
Bei der Bestimmung wird häufig die Titration einer Ramie-Lösung (normalerweise HClO 4) in einem nichtwässrigen Medium verwendet. Mit dieser Methode können Sie häufig die Organisation separat bestimmen. und Nicht-org. Basen in Mischungen, sowie org. Basen unterschiedlicher Stärke, wenn sie zusammen vorhanden sind. Amine können wie Hydroxyderivate durch das Verhältnis ihrer Acylierung bestimmt werden. Zur Bestimmung primärer Aromaten. Amine werden oft mit Lösung in einem sauren Medium titriert, begleitet von der Bildung einer Diazoverbindung. Eine ähnliche Titration sekundärer Amine führt zu deren N-Nitrosierung und wird auch in der Analyse verwendet. Bei der Mikroanalyse von Primäraromaten. Amine werden die resultierenden Diazoverbindungen üblicherweise mit den entsprechenden Azokomponenten kombiniert und der resultierende Farbstoff spektrophotometrisch bestimmt. Bei der Analyse von Gemischen aus primären, sekundären und tertiären Aminen wird am häufigsten die Titration mit HClO 4 -Lösung in einem nichtwässrigen Medium der Ausgangsmischung (alle Amine werden titriert), der Mischung nach Acetylierung mit Essigsäureanhydrid (nur tertiär) verwendet Amine werden titriert) und die Mischung nach Behandlung mit Acetylaceton oder Salicylaldehyd (die Summe aus sekundären und tertiären Aminen wird titriert).
Um Aryldiazoniumsalze mit einer Lösung der analysierten Substanz zu bestimmen, titrieren Sie abgewogene Portionen der Azokomponente (3-Methyl-1-phenyl-5-pyrazolon, m-Phenylendiamin usw.) oder geben Sie eine Lösung der Azokomponente zur analysierten Substanz Der Schwarm wird mit NaNO 2 -Lösung im sauren Milieu titriert. Bei der Analyse von Diazoverbindungen kann auch die Gasometrie eingesetzt werden. Analyse basierend auf der Zersetzung der untersuchten Verbindung. unter Freisetzung von N 2, dessen Volumen gemessen wird. Manchmal, wie im Fall der Analyse von Aminen, werden Diazoverbindungen durch die Kombination mit letzteren bestimmt. spektrophotometrisch reich. Bestimmung des resultierenden Farbstoffs.
Hydrazine werden üblicherweise iodometrisch titriert. Im Falle von Thiolen kann auch Wechselwirkung genutzt werden. sie mit Silbersalzen oder Säure-Base-Titration. Org. Sulfide werden mit einem Bromid-Bromat-Gemisch oxidiert, der Überschuss wird titrimetrisch bestimmt.
Weit verbreitet für Qualität. und Mengen. funktionell Selektive und recht empfindliche Methoden der IR-Spektroskopie und NMR wurden ebenfalls analysiert.
Die Entstehung der stereospezifischen Analyse von org. in-in der 2. Hälfte. 20. Jahrhundert mit der Entwicklung der Chromatographie verbunden Methoden. Zur Trennung von Enantiomeren wird meist eine Vorreaktion zwischen den analysierten Substanzen und optisch aktiven Reagenzien zur Bildung von Diastereomeren durchgeführt, die dann durch Gas-Flüssigkeits- oder Hochleistungsflüssigkeitschromatographie auf Säulen mit optisch aktiven stationären Phasen getrennt werden.
Molekulare Analyse von org. in-in gegründet ch. arr. über den Einsatz von Chromatographie und anderen. Spektralmethoden, die es ermöglichen, die Struktur der Organisation zu ermitteln. Verbindungen.
Phasenanalyse, die eine qualitative und quantitative Analyse kristalliner Materialien ermöglicht. org-Formulare Verbindung, durchgeführt mit Radiographie Und Elektronographie. Röntgen, Strukturanalyse ermöglicht es Ihnen, die Strukturstruktur der Organisation mit hoher Genauigkeit festzulegen. v-va, bestimmen Sie die Längen der Bindungen zwischen Atomen und die Winkel zwischen den Bindungen.
Die oben aufgeführten Analysemethoden basieren auf der direkten Bestimmung der analysierten Substanzen oder daraus gewonnener Derivate. In O. v. A. Auch indirekte Methoden kommen häufig zum Einsatz. So können beispielsweise Kohlenstoffverbindungen in Form von schwerlöslichem Silber oder anderen Salzen aus dem analysierten Gemisch isoliert und anschließend mithilfe der Atomabsorptionsmethode verwendet werden. Spektroskopie oder Röntgenfluoreszenzanalyse zur Bestimmung der Menge des entsprechenden Metalls; Basierend auf den Ergebnissen einer solchen Analyse kann der Kohlendioxidgehalt berechnet werden. Bei der Flüssigkeitschromatographie ist die indirekte Detektion der getrennten Substanzen wirksam, bei der der mobilen Phase eine aktive Komponente zugesetzt wird, die mit den Trennprodukten oder den zu chromatographierenden Substanzen leicht nachweisbare Verbindungen bildet.
Die Analysemethoden und die verwendeten Geräte richten sich nach der konkreten Aufgabenstellung von O. v. a.: Bestimmung des Hauptbestandteils der Mischung, org. oder nicht-org. Verunreinigungen in org. Wow, Org. Verunreinigungen in anorganischen in-ve oder Analyse einer komplexen Mehrkomponentenmischung von in-in.
Methoden O. Jahrhundert. A. weit verbreitet in der Entwicklung der Industrietechnologie. produziert von org. Produkte und im Produktionsprozess selbst für die Entwicklung von Methoden zur Analyse von Rohstoffen, Hilfsstoffen. dazwischen, dazwischen. Produkte in verschiedenen Produktionsstadien, um die Produktion zu steuern. Prozess, Fertigprodukte, Abwasser und Gasemissionen, zur Identifizierung von Verunreinigungen in Zwischen- und Endprodukten und zur Entwicklung von Analyten. Techniken, die die nötige Kinetik gewährleisten. Forschung. In jedem Fall ist es notwendig, das optimale auszuwählen. Optionen für Analysemethoden und deren Kombinationen gemäß den Anforderungen an Geschwindigkeit, Reproduzierbarkeit, Genauigkeit usw.
Bei der Entwicklung eines Analyten. Teile des normativen und technischen. Dokumentation für Rohstoffe, Hilfsstoffe. Materialien und Fertigprodukte ermitteln zunächst die minimal notwendige und ausreichende Anzahl an Analyten. Indikatoren. Zu diesen Indikatoren gehören Schmelzpunkt, pH-Wert und Basengehalt. Stoffe im Produkt, die durch eine direkte Methode (meist titrimetrisch mittels Potentiometrie) oder indirekt bestimmt werden, indem von der Masse des gesamten Produkts die durch Chromatographie bestimmte Masse der Verunreinigungen abgezogen wird. (am häufigsten), elektrochemisch. oder spektrophotometrisch. Methoden. Bei Verwendung der Funktion Analyse zur Bestimmung der Hauptsache Artikel wählen normalerweise eine Methode, die die Bestimmung dieses Artikels anhand der Funktion beinhaltet. Gruppe, die in der letzten Phase ihres Empfangs gebildet wurde. Wenn der analysierte Stoff durch mehrstufige Synthese gewonnen wird, wird er gegebenenfalls nach verschiedenen Funktionen bestimmt. Gruppen. Analytiker. Die für die Analyse von Rohstoffen und Fertigprodukten gewählten Methoden müssen Kap. arr. gute Reproduzierbarkeit und Genauigkeit.
In der Produktionskontrolle verwendete Analysemethoden müssen schnell und kontinuierlich sein (z. B. Redoxmetrie, pH-Metrie usw.). Die Grundlage der Methoden zur Überwachung von Produktionsprozessen ist org. in-in liegt oft die Definition einer verschwindenden Funktion. Gruppe, d. h. eine Gruppe, die in einer bestimmten Produktionsphase eine Transformation durchläuft, was es ermöglicht, das Ende der entsprechenden Phase genau zu erfassen. In diesem Fall werden häufig Dünnschicht-, Gas-Flüssigkeits-, Hochleistungsflüssigkeitschromatographie, Spektrophotometrie und Elektrochemie eingesetzt. Methoden, Fließinjektion. Analyse.
Zur Analyse wird es Intervalle geben. Die Titrimetrie wird am häufigsten für hergestellte Produkte und zur Reaktionsanalyse eingesetzt. Gemische-komplexe Chromatographie. und Spektralmethoden, einschließlich Gaschromatographie-Massenspektrometrie, einer Kombination aus Gaschromatographie und Fourier-Transformations-Infrarotspektroskopie.
Die Analyse von Umweltobjekten hat große Bedeutung erlangt. Bei der Entwicklung geeigneter Methoden zur Analyse grundlegender Anforderungen an sie sind eine hohe Empfindlichkeit und eine korrekte Identifizierung der zu bestimmenden Stoffe. Diese Anforderungen werden durch Gaschromatographie-Massenspektrometrie unter Verwendung von zwei oder mehr stationären Phasen erfüllt.
Im klinischen Bereich Bei der Analyse (Analyse von Blut, Urin, Gewebe und anderen Objekten auf den Gehalt an Arzneimitteln, Metaboliten, Steroiden, Aminosäuren usw.) ist nicht nur die Empfindlichkeit, Genauigkeit und Schnelligkeit der Analyse wichtig, sondern auch die Reproduzierbarkeit ihrer Ergebnisse. Wenn die letztgenannte Anforderung kritisch ist, werden Gaschromatographie-Massenspektrometrie unter Standardbedingungen sowie Hochdurchsatz-Flussinjektion eingesetzt. Analyse und eine Vielzahl von Enzymmethoden mit hoher Selektivität.
Zündete.: Guben Weil, Methoden der organischen Chemie, Bd. 2, Methoden der Analyse, trans. mit ihm. 4. Aufl., M.. 1963; Cheronis N. D., Ma T. S., Mikro- und Halbmikromethoden der organischen Funktionsanalyse, trans. aus Englisch, M., 1973; Siggia S.. Hannah J. G., Quantitative organische Analyse durch funktionelle Gruppen, trans. aus dem Englischen, M.; 1983. B. ICH. Kolokolow.
Chemische Enzyklopädie. - M.: Sowjetische Enzyklopädie. Ed. I. L. Knunyants. 1988 .
Sehen Sie, was „ANALYSE ORGANISCHER STOFFE“ in anderen Wörterbüchern ist:
Die Wasseranalyse ist eine Methode zur Untersuchung der Eigenschaften und Qualitäten von Wasser. Es wird verwendet, um die Menge verschiedener Substanzen im Wasser zu bestimmen, die für industrielle und häusliche Zwecke oder für wissenschaftliche Zwecke mit Menschen in Kontakt kommen. Inhalt 1 Wasserarten für ... ... Wikipedia
Bei der Bodenanalyse handelt es sich um eine Reihe von Vorgängen zur Bestimmung der Zusammensetzung sowie der physikalisch-mechanischen, physikalisch-chemischen, chemischen, agrochemischen und biologischen Eigenschaften des Bodens. Führen Sie mechanische (granulometrische), chemische, ... ... Wikipedia durch
WASSERANALYSE- wird durchgeführt, um die Qualität des Wassers zu bestimmen und die Möglichkeit seiner Nutzung zur Versorgung von Fischteichen zu ermitteln. Ein V. findet viermal im Jahr statt: im Frühling (während des Frühjahrshochwassers), im Hochsommer (Juli), im Herbst (während des Herbstes... ... Teichfischzucht
Analyse- ANALYSE (von griech. Analyse, Zerlegung, Zerstückelung) ist das Verfahren zur realen oder mentalen Aufteilung eines Objekts, Phänomens oder Prozesses sowie deren Beziehungen in Komponenten, Elemente, Eigenschaften, Funktionen und Subsysteme. Das Verfahren... ... Enzyklopädie der Erkenntnistheorie und Wissenschaftstheorie
Identifizierung (Nachweis) der analysierten Bestandteile und ungefähren Mengen, Beurteilung ihres Gehalts in Gewässern und Materialien. Als Komponenten kann es sein Atome und Ionen, Isotope von Elementen und einzelne Nuklide, Moleküle, funktionell. Gruppen und Radikale... Chemische Enzyklopädie
Bestimmung von Inhalten (Masse, Konzentration etc.) oder Mengen. Verhältnisse der Komponenten in der analysierten Probe. Bestimmte Komponenten können sein. Atome, Moleküle, Isotope, funktionell. Gruppen, Phasen usw. (siehe Elementaranalyse, Molekulare ... ... Chemische Enzyklopädie
Bildungseinrichtung „Brest State University benannt nach A.S. Puschkin“
Institut für Chemie
KURSARBEIT
Methoden zur Untersuchung organischer Verbindungen
Durchgeführt:
Student im 5. Jahr,
Fakultät für Biologie
Fachgebiet „Biologie. Chemie"
Vollzeitausbildung
Petruchik Irina Alexandrowna
Wissenschaftlicher Leiter:
Borichevsky
Alexander Iwanowitsch
Brest, 2012
Methoden zur Untersuchung organischer Verbindungen
INHALTSVERZEICHNIS
EINFÜHRUNG……………………………………………………………….. 3
- Klassifizierung von Methoden zur Untersuchung organischer Substanzen………. 4
Die einfachsten Methoden zur Untersuchung organischer Substanzen
2.1.1 Kristallisation………………………………………… ……… 6
2.1.2 Sublimation…………………………………………………………. 7
2.1.3 Destillation……………………………………………………….. 8
2.1.4 Chromatographie…………………………………………… …. 9-11
2.2 Analyse organischer Stoffe………………………………….. 12-13
- Physikalisch-chemische Methoden zur Untersuchung organischer Substanzen... 14
3.2 Kalorimetrie……………………………………………………………… ……… 17
3.3 Röntgen- und Elektronenbeugung…………………………… 18-19
3.4 Elektrochemische Forschungsmethoden…………………… 20-21
3.5 Spektroskopie…………………………………………… …….. 22-27
ABSCHLUSS…………………………………………………… ……….…. 28
REFERENZENLISTE…………………………. 29
EINFÜHRUNG
Die Untersuchung organischer Substanzen zielt darauf ab, die Struktur der Substanz, ihre räumliche Struktur und charakteristische Molekülorbitale zu ermitteln, die Wechselwirkung von Atomen und Molekülen zu untersuchen und die Geschwindigkeiten und Mechanismen von Reaktionen zu untersuchen. Aufgrund der großen Anzahl verschiedener organischer Verbindungen ist es unmöglich, ein einziges Analyseschema zu entwickeln, wie dies häufig bei der anorganischen quantitativen Analyse der Fall ist. Und doch ist es durch systematische Forschung möglich, organisches Material recht zuverlässig und schnell zu identifizieren.
Unabhängig von der Forschungsmethode ist die Aufklärung der Struktur organischer Materie das Hauptziel ihrer Forschung. Die mit der Erforschung der einen oder anderen organischen Verbindung verbundenen Interessen sind jedoch bereits anderer Natur. Von besonderer Bedeutung sind Fragen im Zusammenhang mit den natürlichen Ressourcen unseres Planeten. Wir wissen, dass Öl- und Gasquellen für die Menschheit von besonderer Bedeutung sind, aber sie sind begrenzt. Daher ist das Problem der Suche nach neuen Rohstoffen für die organische und petrochemische Synthese sowie die künstliche Förderung von Öl und Gas dringlich geworden. Dies ist jedoch nur einer der Gründe für die Untersuchung organischer Materie. Wenn Sie sich umschauen, ist alles Leben auf der Erde organische Chemie. Dementsprechend ist die Erforschung organischer Substanzen der Schlüssel zu globalen Entdeckungen auf dem Gebiet der belebten Natur, die Möglichkeit, alle lebenswichtigen Prozesse zu erlernen, Wege zu finden, viele schreckliche Krankheiten zu heilen, selbst lebende Materie zu erschaffen usw.
- Klassifizierung von Methoden zur Untersuchung organischer Substanzen.
- die einfachsten Untersuchungsmethoden: Reinigung organischer Substanzen (Kristallisation, Sublimation, Destillation, Chromatographie, Gelfiltration, Elektrophorese) und Analyse organischer Substanzen (quantitative und qualitative Elementaranalysen);
- physikalische und chemische Methoden: Refraktometrie, Kalorimetrie, Messung elektrischer Dipolmomente, Radiographie und Elektronenbeugung, elektrochemische Methoden (Polarographie, anodische Voltammetrie), Spektroskopie (Photoelektronen-, Massenspektroskopie, Infrarot usw.)
Die einfachsten Methoden zur Untersuchung organischer Substanzen
- Organische Reinigung
Um die Reinheit eines Stoffes zu charakterisieren, werden folgende Konstanten und Methoden verwendet: Schmelzpunkt, Kristallisationstemperatur, Siedepunkt, Lichtbrechungsindex, Dichte, Absorptionsspektrendaten (Absorptionsintensitätskoeffizient in elektronischen und Infrarotspektren), Kernspinresonanz (NMR). ) Spektrendaten, Massenspektrometrie, chromatographische Analyse, Lumineszenzanalyse usw.
Einen reinen Stoff zu erhalten bedeutet, ein gegebenes Stoffgemisch in einzelne Stoffe aufzutrennen und diese bis zum gewünschten Reinheitsgrad zu reinigen. Hierbei ist zwischen zwei Methodengruppen zu unterscheiden: Methoden zur Auftrennung eines Gemisches in noch nicht reine Komponenten und Methoden zur Endreinigung.
Wenn es um die Reinheit chemischer Stoffe geht, muss man sich darüber im Klaren sein, dass man sich einen absolut reinen Stoff nur theoretisch vorstellen kann. Es gibt keine absolut reinen Stoffe und kann es auch nicht geben. Je nach Reinigungsmethode enthält der Stoff eine gewisse Menge an Verunreinigungen. Mit herkömmlichen Reinigungsmethoden kann ein Gehalt der Hauptsubstanz von 99,9...99,95 % erreicht werden. Durch spezielle Tiefenreinigungsverfahren ist es möglich, den Verunreinigungsgehalt organischer Stoffe auf 10 -3 ....10 -4 % zu reduzieren.
2.1.1 Kristallisation
Die Kristallisation ist eine klassische Methode zur Reinigung kristalliner Stoffe. Die Methode basiert auf der Tatsache, dass verschiedene Stoffe in einem bestimmten Lösungsmittel unterschiedliche Löslichkeiten haben und eine Temperaturabnahme (mit seltenen Ausnahmen) zu einer Abnahme der Löslichkeit von Stoffen führt. Durch Filtrieren der heißen Lösung werden unlösliche Verunreinigungen abgetrennt und nach dem Abkühlen wird der Stoff in Form von Kristallen aus der Lösung freigesetzt. Wiederholte Umkristallisationen verringern in der Regel die Menge an Verunreinigungen. Eine Variante des Verfahrens ist die Schmelzkristallisation. Eine besondere Option – das Zonenschmelzen – dient der Tiefenreinigung von Stoffen.
Zum Beispiel: Wir müssen Salicylsäure von Verunreinigungen reinigen. Dazu nehmen wir eine vorgewogene Masse dieser Säure und berechnen das erforderliche Lösungsmittelvolumen – Wasser, um eine gesättigte Lösung zu erhalten, die anschließend kristallisiert werden kann.
2.1.2 Sublimation (Sublimation)
Viele kristalline Stoffe besitzen die Fähigkeit zu sublimieren, d.h. bis zum Übergang in die Gasphase unter Umgehung der flüssigen Phase, gefolgt von der Kristallisation aus der Gasphase. Mit dieser Methode können Sie sublimierende Stoffe von nicht sublimierenden Verunreinigungen trennen und ein Stoffgemisch mit unterschiedlichen Sublimationstemperaturen bzw. Kristallisationstemperaturen aus der Gasphase trennen (Gradientensublimation). Wenn Stoffe bei hohen Temperaturen schwer zu sublimieren und zu zersetzen sind, kommt die Sublimation im Vakuum oder Hochvakuum zum Einsatz – bis 0,0013 Pa (10 -5 mm Hg; 1 mm Hg = 133,3 Pa). Zur Tiefenreinigung kommt die Hochvakuumsublimation in verschiedenen Ausführungen zum Einsatz.
Die Reinigung eines Feststoffs durch Sublimation ist nur möglich, wenn sein Dampfdruck höher ist als der Dampfdruck der Verunreinigungen. Wenn der Dampfdruck des Feststoffs dem angelegten Druck entspricht, werden die besten Ergebnisse erzielt.
Beispiel: E-Stilben wird bei einer Temperatur von 100 °C und einem Druck von 20 mm Hg sublimiert. Kunst.
2.1.3 Destillation (Destillation)
Für viele niedrig schmelzende Stoffe und die meisten Flüssigkeiten ist eine gute Reinigungsmethode geeignet
Fraktionierte Destillation, sofern der Siedepunktunterschied der Gemischbestandteile ausreichend groß ist und keine azeotropen Gemische entstehen. Die Selektivität (Effizienz) der fraktionierten Destillation kann durch spezielle Geräte erhöht werden: Rückflusskühler, Destillationskolonnen etc. Bei hochsiedenden Stoffen kommt die Vakuumdestillation zum Einsatz. Eine Variante des Verfahrens ist die Destillation von Zweikomponentensystemen, die sich beim Abkühlen trennen, zum Beispiel die Wasserdampfdestillation: Limonen (Siedepunkt 178 °C bei 760 mm Hg) wird mit Wasser (Siedepunkt 100 °C bei 760 mm Hg) destilliert .) bei einer Temperatur von 98 o C. In diesem Fall beträgt das Mengenverhältnis im Destillat (in Gramm) Limonen:Wasser 1:1,54.
2.1.4 Chromatographie
Chromatographische Trennverfahren basieren auf der unterschiedlichen Fähigkeit von Substanzen, an der Oberfläche des Sorbens zu adsorbieren oder sich zwischen zwei nicht mischbaren Phasen (Flüssigkeit-Flüssigkeit, Flüssigkeit-Gas) zu verteilen, von denen sich eine Phase (Flüssigkeit) auf der Oberfläche des Sorbens befindet Sorptionsmittel. Daher gibt es verschiedene Arten der Chromatographie, nämlich: Flüssigkeitsadsorptions- und Verteilungschromatographie, Gaschromatographie.
Die Flüssibasiert auf der unterschiedlichen Fähigkeit von Substanzen, an der Oberfläche des Sorbens zu sorbieren und beim Passieren eines Lösungsmittels – Eluent – zu desorbieren. Als Sorbentien werden Aluminiumoxid, Kieselsäure und Siliziumdioxid (Kieselgele), körnige Polysaccharide (Dextrane) oder andere Polymere verwendet, die in einem Lösungsmittel quellen und ein körniges Gel bilden (Gelchromatographie).
Die Flüssiist eine Art der Adsorptionschromatographie, bei der das Sorptionsmittel (Träger) mit einem dünnen Flüssigkeitsfilm überzogen wird. Das Elutionsmittel ist in der Regel ein Lösungsmittel, das sich nicht mit der Flüssigkeit auf dem Sorbens vermischt. Beim Durchleiten des Eluenten werden Stoffe zwischen der flüssigen Phase und dem Eluenten verteilt. Diese Art der Chromatographie eignet sich am besten zur Trennung von Substanzen, die gut wasserlöslich sind oder wasserlösliche Salze bilden können. Zu diesen Substanzen zählen Zucker, Aminosäuren, viele organische Farbstoffe, die meisten Alkaloide, Mono- und Polycarbonsäuren, Alkohole usw.
Ein Beispiel für die Flüssigkeitschromatographie einer Mischung aus synthetischen Phospholipid-Standards (1) und einer Probe eines rohen Lipidextrakts aus der Zellmembran menschlicher Erythrozyten (2) auf einer Normalphasensäule bei Detektion mit einem Laserlichtstreuungsdetektor.NL – neutral Lipide; PE – Phosphatidylethanolamin; PS – Phosphatidylserin; PC – Phosphatidylcholin; SM – Sphingomyelin.
Die Gaschromatographie dient der Trennung von Gemischen aus gasförmigen oder leichtflüchtigen flüssigen und festen Stoffen. Das Prinzip der Methode ähnelt der Flüssigkeitschromatographie. Das zu trennende Gemisch wird mit einem Trägergas (H 2, N 2, He) verdünnt und in Adsorptionskolonnen eingeleitet. Das Trägergas ist sowohl Lösungsmittel als auch Eluent. Als Sorbentien werden feine Pulver aus Silikatmaterialien verwendet, die rein (Gasadsorptionschromatographie) oder mit einem Film aus nichtflüchtiger Flüssigkeit überzogen (Gas-Flüssigkeits-Chromatographie) sein können. Es werden auch Kapillaren verwendet, die innen mit einem Film aus nichtflüchtiger Flüssigkeit beschichtet sind (Kapillarchromatographie). Das Trägergas desorbiert nach und nach die Gemischbestandteile und reißt sie mit. Das Vorhandensein organischer Substanzen im Trägergas und deren Menge wird mit speziellen Detektoren erfasst und von einem Schreiber aufgezeichnet. Bei der präparativen Chromatographie wird das Trägergas anschließend durch spezielle Vorlagen geleitet, in denen organische Substanzen durch Gefrieren eingefangen werden.
Mit dieser Methode kann eine vollständige Trennung der Mischung erreicht werden. Beim Einsatz von Hochleistungsadsorptionssäulen wird das Verfahren als präparative Methode zur Trennung kleiner Stoffmengen (1...10 g) eingesetzt.
Beispiel Gaschromatographie: Hochgeschwindigkeitsanalyse explosiver Dämpfe an einer Polykapillarsäule bei einer Temperatur von 170 °C.
Mit einer Polykapillarsäule mit einer Länge von nur 22 cm können Sie Spuren explosiver Dämpfe in 2,5 Minuten erkennen und identifizieren: 1 - 2,6-Dinitrotoluol, 2 - 2,4-Dinitrotoluol. 3 - 2,4,6-Trinitrotoluol, 4 - 3,4,5-Trinitrotoluol, 5 - 2,3,4-Trinitrotoluol, 6 - Hexogen. 7 - Tetryl.
- Analyse organischer Stoffe
Die erste Aufgabe besteht in der qualitativen und quantitativen Bestimmung der Elementzusammensetzung. Anschließend wird anhand der Elementaranalysedaten die einfachste Summenformel berechnet, das Molekulargewicht bestimmt und die wahre molekulare Bruttoformel berechnet. Der letzte Schritt besteht schließlich darin, die Molekülstruktur zu bestimmen. Zu diesem Zweck werden chemische Methoden (schrittweise Spaltung, Ableitung) und in jüngster Zeit zunehmend physikalisch-chemische Methoden (Massenspektroskopie, Röntgenbeugungsanalyse, Spektroskopie) eingesetzt.
Quantitative und qualitative Elementaranalyse
Die Analysemethoden basieren auf dem vollständigen Abbau organischer Stoffe durch Oxidation oder auf andere Weise und der Bestimmung chemischer Elemente mit bekannten Methoden. Kohlenstoff wird in Form von CO 2 bestimmt, Wasserstoff – in Form von H 2 O, Stickstoff – durch Messung des Volumens von N 2 oder Bestimmung von NH 3 oder NaCN (je nach Art der Spaltung), Halogene – in Form von Halogenidionen, Schwefel – in Form von Sulfat- oder Sulfidionen, Phosphor bedeutet Phosphation usw.
Kohlenstoff und Wasserstoff werden durch Erhitzen mit CuO qualitativ bestimmt:
C n H 2n +3nCuO>nCO 2 +nH 2 O+3nCu
Und das freigesetzte Kohlenmonoxid wird durch Einleiten des Gases in die Ba(OH) 2-Lösung nachgewiesen, und Wasser wird visuell an den Wänden des Reagenzglases nachgewiesen.
Stickstoff, Schwefel und Halogene werden qualitativ durch Fusion mit Natrium bestimmt. Die resultierenden NaCN-, Na 2 S- und Natriumhalogenide werden in wässriger Lösung durch herkömmliche analytische Reaktionen nachgewiesen.
Für die quantitative Analyse organischer Verbindungen gibt es spezielle Proben. Bisher wurden meist Anlagen zur Makroanalyse eingesetzt (Probengewicht 0,2 ... 0,5 g). Heutzutage sind verschiedene Geräte für die Mikroanalyse (Probe 0,001 ... 0,01 g), für die Ultramikroanalyse (Probe 10 -5 ... 10 -4 g) üblich. Zur quantitativen Bestimmung von Kohlenstoff und Wasserstoff werden Geräte eingesetzt, bei denen organische Stoffe im Sauerstoffstrom verbrannt werden: CO 2 wird mit einer KOH-Lösung, H 2 O mit einem speziellen Absorptionsmittel aufgefangen und durch Wiegen bestimmt. Zur Quantifizierung von Stickstoff wird der Stoff beim Erhitzen mit CuO verbrannt und das freigesetzte Gasvolumen in einem Azometer über einer KOH-Lösung gemessen. Halogene und Schwefel werden quantifiziert, indem die Probe in einer Sauerstoffatmosphäre verbrannt, die Gase in Wasser gelöst und die Halogenidionen oder Sulfationen titriert werden.
Automatische Mikroanalysatoren wurden nach dem Prinzip der Gaschromatographie entwickelt, bei der gleichzeitig Kohlenstoff, Wasserstoff, Stickstoff und Schwefel bestimmt werden.
Das Molekulargewicht einer Verbindung wird üblicherweise durch Massenspektrometrie bestimmt.
- Physikalisch-chemische Methoden zur Untersuchung organischer Substanzen
- Spektrale und andere optische Methoden;
Elektrochemische Methoden;
Chromatographische Analysemethoden.
Eine Gruppe elektrochemischer Analysemethoden, die auf der Messung elektrischer Leitfähigkeit, Potentiale und anderer Eigenschaften basieren, umfasst Methoden der Konduktometrie, Potentiometrie, Voltammetrie usw.
Um jedoch die bessere Effizienz dieser Methoden und ihre tatsächlich große praktische Bedeutung genau zu überprüfen, betrachten wir zum Vergleich andere physikalisch-chemische Methoden.
- Refraktometrie
, wobei n der Brechungsindex des Lichts für die Natrium-D-Linie (589 nm) ist; M ist das Molekulargewicht der Substanz; ?? - Dichte.
Die molekulare Brechung hat additive Eigenschaften, d.h. Die molekulare Brechung eines Moleküls kann durch Summieren der Brechungen der Bestandteile des Moleküls ermittelt werden. Solche Komponenten sind chemische Bindungen und eine Reihe von Bindungen und Atomen. Diese Brechungen werden aus Untersuchungen vieler organischer Verbindungen berechnet und sind in Nachschlagewerken zu finden. Zum Beispiel:
R CH4 = 4 R C-H ; R CH3NO2 = 3 R C-H +R C-N +R NO2
Das Phänomen der Lichtbrechung hängt mit der Polarisierbarkeit des elektronischen Molekülsystems zusammen. Unter dem Einfluss des elektromagnetischen Lichtfeldes kommt es zur Polarisation von Molekülen, hauptsächlich ihrer elektronischen Systeme. Je beweglicher das elektronische System eines Moleküls ist, desto größer ist der Brechungsindex des Lichts und die Molekülbrechung.
Molekulare Brechungsstudien können verwendet werden, um die Struktur einer Verbindung zu bestimmen. Daher wird für die untersuchte Verbindung die molekulare Brechung experimentell bestimmt und mit der Brechung verglichen, die durch Summieren der Brechungen der Bindungen gemäß der vorgeschlagenen Strukturformel erhalten wird. Wenn die Ergebnisse übereinstimmen, kann die Struktur als bewiesen gelten; wenn nicht, müssen wir nach einer anderen Struktur suchen. In einigen Fällen wird ein starker Anstieg der molekularen Brechung im Vergleich zu erwartet beobachtet (Brechungserhöhung). Dies ist typisch für gekoppelte Systeme.
Anhand der molekularen Brechungswerte chemischer Bindungen, Atome, Moleküle und Ionen lässt sich deren Polarisierbarkeit qualitativ beurteilen. Die Polarisierbarkeit eines Moleküls (Ion, Bindung) ist seine Fähigkeit zur Polarisation, d.h. zu einer Änderung der Position der Kerne und des Zustands der Elektronenwolke unter dem Einfluss eines externen elektrischen Feldes. Es kommt hauptsächlich zu elektronischer Polarisation.
3.2 Kalorimetrie
Kalorimetrie ist eine Methode zur Untersuchung der thermischen Auswirkungen chemischer Reaktionen und Phasenübergangsprozesse (z. B. Schmelzen, Kristallisation, Sublimation, Kondensation). Der Prozess (Reaktion) wird in speziellen Geräten – Kalorimetern – durchgeführt und die freigesetzte oder aufgenommene Wärme quantifiziert.
Die molaren Verbrennungswärmen von Stoffen werden kalorimetrisch bestimmt. Aus der Verbrennungswärme (W) wiederum wird die Bildungswärme eines Stoffes E bzw. die Standardbildungsenthalpie ?H 0 berechnet. Die Bildungswärme eines Stoffes kann anhand von Elementen im atomaren Zustand oder von Elementen im „Standard“-Zustand (Kohlenstoff in Form von Graphit, Wasserstoffgas usw.) berechnet werden, und die resultierenden Zahlenwerte natürlich: abweichen. Bei der Betrachtung tabellarischer Daten müssen Sie hierauf besonders achten. Typischerweise werden die Bildungswärmen von Stoffen für einen Prozess aus den Atomen der Elemente und ?H 0 – aus den Elementen im „Standard“-Zustand berechnet. Zum Beispiel die Bildungswärme von Kohlenwasserstoffen aus Atomen:
- nS - ] – W, wobei W die Verbrennungswärme ist; - Bildungswärme von CO 2 (393,5 kJ/mol); - Bildungswärme von Wasser (285,8 kJ/mol); S – Zerstäubungswärme (Sublimation) von Kohlenstoff (Graphit) (-715 kJ/mol); - Zerstäubungswärme (Dissoziation) eines Wasserstoffmoleküls (-436 kJ/mol).
Je niedriger die Verbrennungswärme ist, desto größer ist die Bildungswärme von Verbindungen gleicher Zusammensetzung.
Diese Methode dient hauptsächlich dem Vergleich und der Charakterisierung der Stabilität und Reaktivität organischer Verbindungen.
3.3 Radiographie und Elektronenbeugung
Die Röntgenmethode – Röntgenbeugungsanalyse – basiert auf der Beugung von Röntgenstrahlen in einem Kristall einer Substanz. Röntgenstrahlen (elektromagnetische Strahlung mit einer Wellenlänge von 0,1-10 nm) interagieren beim Durchgang durch einen Kristall mit den elektronischen Hüllen von Atomen. Als Ergebnis dieser Wechselwirkung kommt es zur Beugung von Röntgenstrahlen und auf dem fotografischen Film entsteht ein Beugungsmuster – Punkte oder Kreise. Aus dem Beugungsmuster werden mit Hilfe komplexer Berechnungen Informationen über die Platzierung von Molekülen in der Elementarzelle eines Kristalls sowie über die Abstände zwischen Atomen und die Winkel zwischen chemischen Bindungen gewonnen. Je kleiner die Anzahl der Elektronen in einem Atom ist, desto schwächer sind die Röntgenreflexionen. Daher ist es sehr schwierig, die Position von Wasserstoffatomen zu bestimmen.
Die Elektronenbeugungsmethode ähnelt der Röntgenmethode und basiert auf der Wechselwirkung eines Elektronenflusses mit einer Substanz. Der durch eine Substanz fließende Elektronenfluss ähnelt elektromagnetischer Strahlung mit einer sehr kurzen Wellenlänge und erzeugt ein Beugungsmuster. Diese Beugungsmuster (Elektronenbeugungsmuster) können für Stoffe im gasförmigen Zustand oder für sehr dünne Filme erhalten werden. Elektronenbeugung entsteht durch die Wechselwirkung von Elektronen mit Atomkernen.
Diese Methoden der Strukturanalyse ermöglichen es, die vollständige Struktur des Moleküls zu bestimmen – interatomare Abstände, Winkel zwischen Bindungen, d.h. die genaue räumliche Anordnung aller Atome eines Moleküls in einem Kristallgitter oder im gasförmigen Zustand. Die Struktur so komplexer Naturstoffe wie Saccharose, Penicillin, Strychnin, Vitamin B 12, einiger Proteine (Myoglobin) und Nukleinsäuren wurde mittels Röntgenbeugungsanalyse bestimmt.
Durch Röntgenforschungsmethoden wurde festgestellt, dass der kovalente Radius von Atomen während der sp 2 - und sp-Hybridisierung je nach Bindungstyp variiert, beispielsweise bei der Doppelbindung C=C (C sp2 - C sp2), Der kovalente Radius des Kohlenstoffatoms C sp2 ist kleiner als bei =C-C-Bindungen (C sp2 - C sp3). Im 1. Fall sind es 0,067 nm, im 2. Fall 0,076 nm und im Fall von Benzol 0,0695 nm, d.h. Die Bindungslänge hängt auch von der Verbindung selbst ab, und für jede Verbindung sind die Bindungslängen ein individuelles Merkmal, das bei der Identifizierung einer bestimmten organischen Verbindung nützlich sein kann.
3.4 Elektrochemische Forschungsmethoden
Elektrochemische Methoden basieren auf der Abhängigkeit der Stromstärke von der angelegten Spannung beim Stromfluss durch eine Lösung in speziell entwickelten Elektrolyseuren. Dadurch entstehen Strom-Spannungs-(Potenzial-)Kurven. Diese Strom-Spannungs-Kurven charakterisieren die an den Elektroden ablaufenden Prozesse. An der Kathode findet eine elektrochemische Reduktion und an der Anode eine elektrochemische Oxidation statt. Abhängig von der Art des untersuchten Prozesses (anodisch oder kathodisch) werden Geräte verwendet, die sich im Verhältnis der Elektrodenflächen, dem Elektrodenmaterial usw. unterscheiden.
Polarographie
Die polarographische Methode basiert auf kathodischen Prozessen (der Zugabe eines Elektrons zu einer Substanz an einer Quecksilbertropfenelektrode). Der Schaltplan eines Polarographen ist sehr einfach. Es besteht aus einer tropfenden Quecksilber-Mikroelektrode mit kontinuierlich erneuerter Oberfläche und einer Referenzelektrode (Quecksilber oder eine andere normale Elektrode). Die Kathodenfläche ist deutlich kleiner als die Anodenfläche, daher sind in diesem Fall die Kathodenpolarisationsvorgänge entscheidend. Die organische Substanz diffundiert zur Kathode, nimmt ein Elektron auf und die Kathode depolarisiert. Die Depolarisation der Kathode beginnt bei einem bestimmten Potential E ext (Reduktions- oder Freisetzungspotential, das für einen bestimmten Depolarisator charakteristisch ist. Infolgedessen beginnt die Elektrolyse und die Stromstärke steigt stark an. Mit einem allmählichen Spannungsanstieg ein bestimmter stationärer Wert der Stromstärke (Grenzstrom) eingestellt, der nicht mehr von der Anstiegsspannung abhängt.
Zur Charakterisierung des Prozesses kann die Polarographie eingesetzt werden:
Die Methode der Polarographie wird häufig zur Bestimmung der Konzentration von Stoffen in Lösungen eingesetzt.
Anodische Voltammetrie
Diese Methode basiert auf anodischen Prozessen (Oxidation einer organischen Verbindung an einer Platin- oder Graphitanode). Aus experimenteller Sicht ähnelt diese Methode der Polarographie.
Anodische Voltammetrie dient der Untersuchung von Oxidationsprozessen:
Die Methode wird auch zur quantitativen Bestimmung von Stoffen in Lösungen eingesetzt.
3.5 Spektroskopie
Spektroskopische Methoden basieren auf der Wechselwirkung eines Stoffes mit elektromagnetischer Strahlung, die zur Absorption oder Emission von Strahlung führt. Wechselwirkungen sind in einem sehr breiten Spektrum elektromagnetischer Wellen möglich, angefangen bei?-Strahlen bis hin zu Radiowellen.
Je nach Bereich des elektromagnetischen Spektrums kommen unterschiedliche experimentelle Methoden und Instrumente zum Einsatz.
Die in der organischen Chemie am häufigsten verwendeten Felder elektromagnetischer Strahlung sind:
- ultravioletter (UV) und sichtbarer Bereich des Spektrums, in dem die Energie absorbiert wird, die zur Anregung von Elektronen im Molekül erforderlich ist (eine Art elektronische Spektroskopie);
- Infrarotbereich (IR), in dem die Energie absorbiert wird, die zur Änderung der Schwingungszustände des Moleküls erforderlich ist (Schwingungsspektroskopie);
- der Bereich der Hochfrequenzstrahlung, in dem Energie aufgewendet wird, um die Spins von Kernen neu auszurichten (kernmagnetische Resonanzspektroskopie – NMR).
Spektrale Methoden werden zur Identifizierung und Strukturaufklärung von Verbindungen, zur Analyse von Gemischen und zur Überwachung des Fortschritts chemischer Umwandlungen eingesetzt. Der Vorteil spektraler Methoden ist der geringe Substanzverbrauch (1 mg oder weniger).
Elektronenspektroskopie
Das elektronische Spektrum entsteht, wenn ein Stoff ultraviolette (Wellenlängen 22–400 nm) und sichtbare (400–800 nm) Strahlung absorbiert. Zwischen diesen Teilen des Spektrums gibt es keinen grundsätzlichen Unterschied; sie unterscheiden sich lediglich dadurch, dass Wellen mit einer Länge von 400-800 nm vom menschlichen Auge wahrgenommen werden und wir die Substanz als farbig wahrnehmen.
Unter dem Einfluss von UV-Licht wird das Molekül angeregt, d.h. Übergang von Elektronen auf ein stärker angeregtes Niveau und Umverteilung der Elektronendichte im Molekül. Die Elektronen, die β-Bindungen bilden, sind am schwierigsten anzuregen; Elektronen von β-Bindungen und freie Elektronenpaare sind leichter anzuregen.
Das Senden Ihrer guten Arbeit an die Wissensdatenbank ist ganz einfach. Nutzen Sie das untenstehende Formular
Studierende, Doktoranden und junge Wissenschaftler, die die Wissensbasis in ihrem Studium und ihrer Arbeit nutzen, werden Ihnen sehr dankbar sein.
Veröffentlicht am http://www.allbest.ru/
Merkmale der Analyse organischer Verbindungen:
Reaktionen mit organischen Stoffen verlaufen langsam unter Bildung von Zwischenprodukten.
Organische Stoffe sind thermolabil und verkohlen beim Erhitzen.
Die pharmazeutische Analyse organischer Arzneistoffe basiert auf den Prinzipien der Funktions- und Elementanalytik.
Funktionsanalyse – Analyse nach funktionellen Gruppen, d.h. Atome, Atomgruppen oder Reaktionszentren, die die physikalischen, chemischen oder pharmakologischen Eigenschaften von Arzneimitteln bestimmen.
Mithilfe der Elementaranalyse wird die Echtheit organischer Arzneimittel überprüft, deren Molekül Atome von Schwefel, Stickstoff, Phosphor, Halogenen, Arsen und Metallen enthält. Die Atome dieser Elemente kommen in elementorganischen Arzneimittelverbindungen in nichtionisiertem Zustand vor; eine notwendige Voraussetzung für die Prüfung ihrer Echtheit ist eine vorläufige Mineralisierung.
Dabei kann es sich um flüssige, feste und gasförmige Stoffe handeln. Gasförmige und flüssige Verbindungen wirken überwiegend narkotisch. Die Wirkung wird durch F – Cl – Br – I abgeschwächt. Jodderivate wirken vor allem antiseptisch. C-F-Verbindung; C-I; C-Br; C-Cl ist kovalent, daher werden für die pharmazeutische Analyse ionische Reaktionen nach der Mineralisierung der Substanz verwendet.
Die Echtheit von Zubereitungen aus flüssigen Halogenkohlenwasserstoffen wird durch physikalische Konstanten (Siedepunkt, Dichte, Löslichkeit) und das Vorhandensein von Halogen bestimmt. Die objektivste Methode besteht darin, die Authentizität anhand der Identität der IR-Spektren des Arzneimittels und der Standardproben festzustellen.
Um das Vorhandensein von Halogenen in einem Molekül nachzuweisen, werden der Beilstein-Test und verschiedene Mineralisierungsmethoden verwendet.
Tabelle 1. Eigenschaften halogenhaltiger Verbindungen
|
Chlorethyl Aethylii cloridum (INN Ethylchlorid) |
Ftorotan 1,1,1-Trifluor-2chlor-2-bromethan (INN Halothan) |
Bromkampfer 3-Brom-1,7,7,trimethylbicycloheptanon-2 |
|
|
Die Flüssigkeit ist transparent, farblos, leicht flüchtig, hat einen eigenartigen Geruch, ist in Wasser schwer löslich und mit Alkohol und Äther in jedem Verhältnis mischbar. |
Die Flüssigkeit ist farblos, transparent, schwer, flüchtig, mit charakteristischem Geruch, schwer löslich in Wasser, mischbar mit Alkohol, Ether und Chloroform. |
Weißes kristallines Pulver oder farblose Kristalle, Geruch und Geschmack, sehr schlecht löslich in Wasser, leicht in Alkohol und Chloroform. |
|
|
Bilignostum pro injectibus Bilignost Bis-(2,4,6-triiod-3-carboxyanilid)-adipinsäure |
Bromiert 2-Bromisovalerianylharnstoff |
||
|
Weißes kristallines Pulver, leicht bitterer Geschmack, praktisch unlöslich in Wasser, Alkohol, Chloroform. |
Weißes kristallines Pulver oder farblose Kristalle mit schwachem spezifischem Geruch, schwer löslich in Wasser, löslich in Alkohol. |
Beilstein-Test
Das Vorhandensein von Halogen wird durch Kalzinieren der Substanz im festen Zustand auf einem Kupferdraht nachgewiesen. In Gegenwart von Halogenen entstehen Kupferhalogenide, die die Flamme grün oder blaugrün färben.
Die Halogene in einem organischen Molekül sind durch eine kovalente Bindung verbunden, deren Stärke von der chemischen Struktur des Halogenderivats abhängt; daher sind verschiedene Bedingungen für die Abspaltung eines Halogens und seine Überführung in einen ionisierten Zustand erforderlich. Die entstehenden Halogenidionen werden durch herkömmliche analytische Reaktionen nachgewiesen.
Chlorethyl
· Mineralisierungsmethode – Kochen mit einer alkoholischen Alkalilösung (aufgrund des niedrigen Siedepunkts erfolgt die Bestimmung unter Rückfluss).
CH 3 CH 2 Cl+KOH c KCl +C 2 H 5 OH
Das entstehende Chloridion wird mit einer Silbernitratlösung durch die Bildung eines weißen, käsigen Niederschlags nachgewiesen.
Сl- + AgNO 3 > AgCl + NO 3 -
Ftorotan
· Mineralisierungsmethode – Fusion mit metallischem Natrium
F 3 C-CHClBr + 5Na + 4H 2 O> 3NaF + NaCl + 2NaBr + 2CO 2
Die entstehenden Chlorid- und Bromidionen werden von einer Silbernitratlösung durch die Bildung weißer, käsiger und gelblicher Niederschläge nachgewiesen.
Fluoridionen werden durch folgende Reaktionen nachgewiesen:
Reaktion mit einer Lösung von Alizarinrot und einer Lösung von Zirkoniumnitrat in Gegenwart von F-, die rote Farbe geht in hellgelb über;
Wechselwirkung mit löslichen Calciumsalzen (es bildet sich ein weißer Niederschlag aus Calciumfluorid);
Verfärbungsreaktion von Eisenthiocyanat (rot).
· Bei Zugabe zu Fluorthankonz. H 2 SO 4, der Wirkstoff befindet sich in der unteren Schicht.
Bromiert
· Mineralisierungsmethode – Kochen mit Alkali (alkalische Hydrolyse in wässriger Lösung), es entsteht der Geruch von Ammoniak:
· Erhitzen mit konz. Schwefelsäure – der Geruch von Isovaleriansäure
Bromkampfer
· Mineralisierungsverfahren nach der R(mit metallischem Zink in alkalischem Medium)
Das Bromidion wird durch Reaktion mit Chloramin B bestimmt.
Bilignost
· Mineralisierungsmethode – Erhitzen mit konzentrierter Schwefelsäure: Es wird das Auftreten violetter Dämpfe von molekularem Jod festgestellt.
· IR-Spektroskopie – eine 0,001 %ige Lösung des Arzneimittels in einer 0,1 N Natronlauge im Bereich von 220 bis 300 nm hat ein Absorptionsmaximum bei l = 236 nm.
Jodoform
Mineralisierungsmethoden:
1) Pyrolyse im trockenen Reagenzglas, violetter Joddampf wird freigesetzt
4CHI 3 + 5O 2 > 6I 2 + 4CO 2 + 2H 2 O
2) Erhitzen mit konz. Schwefelsäure
2CHI 3 + H 2 SO 4 > 3I 2 + 2CO 2 + 2H 2 O + SO 3
Gute Qualität (Reinheit der Halogenkohlenwasserstoffe).
Die Qualität von Chlorethyl und Fluortan wird durch Feststellung des Säuregehalts oder der Alkalität, der Abwesenheit oder des akzeptablen Gehalts an Stabilisatoren (Thymol in Fluortan – 0,01 %), fremder organischer Verunreinigungen, freier Chlorverunreinigungen (Brom in Fluortan), Chloriden, Bromiden und Nicht-Chloriden überprüft. flüchtiger Rückstand.
1) Chlorethyl: 1. Siedepunkt und Dichte bestimmen,
2. Unzulässige Verunreinigung von Ethylalkohol (Reaktion zur Bildung von Jodoform)
2) Bilignost: 1. Erhitzen mit kH 2 SO 4 und Bildung violetter Dämpfe I 2
2. IR-Spektroskopie
3) Ftorotan: 1. IR-Spektroskopie
2. Siedepunkt; Dichte; Brechungsindex
3. Es dürfen keine Cl- und Br-Verunreinigungen vorhanden sein
GF ermöglicht keine quantitative Bestimmung von Chlorethyl, sie kann jedoch durch Argentometrie oder Quecksilbermessung durchgeführt werden.
Die Methode der quantitativen Bestimmung ist die umgekehrte argentometrische Titration nach Volhard nach der Mineralisierung (zur Reaktion siehe Definition der Authentizität).
1. Reaktion vor der Titration:
pharmazeutische medizinische Chlorethyl-Titration
NaBr + AgNO 3 > AgBrv+ NaNO 3
2. Titrationsreaktion:
AgNO 3 + NH 4 SCN > AgSCN v + NH 4 NO 3
3. Am Äquivalenzpunkt:
3NH 4 SCN + Fe(NH 4)(SO 4) 2 >
Die Methode der quantitativen Bestimmung ist die argentometrische Titration nach Kolthoff nach der Mineralisierung (Reaktionen siehe Definition der Authentizität).
1. Reaktion vor der Titration:
3NH 4 SCN + Fe(NH 4)(SO 4) 2 > Fe (SCN) 3 + 2 (NH 4) 2 SO 4
genaue Menge bräunlichrot
2. Titrationsreaktion:
NaBr + AgNO 3 > AgBrv+ NaNO 3
3. Am Äquivalenzpunkt:
AgNO 3 + NH 4 SCN > AgSCNv + NH 4 NO 3
Bleichen
Bilignost
Die Methode der quantitativen Bestimmung ist die indirekte Jodometrie nach oxidativer Spaltung von Bilignost zu Jodat beim Erhitzen mit einer Lösung von Kaliumpermanganat in einem sauren Medium, überschüssiges Kaliumpermanganat wird mit Natriumnitrat entfernt und zur Entfernung überschüssiger salpetriger Säure wird eine Harnstofflösung zugesetzt die Mischung.
Titriermittel – 0,1 mol/l Natriumtitsulfatlösung, Indikator – Stärke, am Äquivalenzpunkt wird das Verschwinden der blauen Farbe der Stärke beobachtet.
Reaktionsschema:
T; KMnO 4 +H 2 SO 4
RI 6 > 12 IO 3 -
Substituentenfreisetzungsreaktion:
KIO 3 + 5KI + 3H 2 SO 4 >3I 2 + 3K 2 SO 4 + 3H 2 O
Titrationsreaktion:
I 2 +2Na 2 S 2 O 3 > 2NaI+Na 2 S 4 O 6
Jodoform
Die Methode der quantitativen Bestimmung ist die umgekehrte argentometrische Titration nach Volhard nach der Mineralisierung.
Mineralisierung:
CHI 3 + 3AgNO 3 + H 2 O> 3AgI + 3HNO 3 + CO 2
Titrationsreaktion:
AgNO 3 + NH 4 SCN > AgSCN v + NH 4 NO 3
Am Äquivalenzpunkt:
3NH 4 SCN + Fe(NH 4)(SO 4) 2 > Fe (SCN) 3 v + 2 (NH 4) 2 SO 4
Lagerung
Chlorethyl in Ampullen an einem kühlen, dunklen Ort, Ftorotan und Bilignost in orangefarbenen Glasflaschen an einem kühlen, trockenen Ort, vor Licht geschützt. Bromkampfer wird in orangefarbenen Glasflaschen an einem kühlen, trockenen Ort aufbewahrt.
Zur Lokalanästhesie wird Chlorethyl, zur Anästhesie Fluorothan verwendet. Bromkampfer wird als Beruhigungsmittel verwendet (manchmal um die Laktation zu stoppen). Bromizoval ist ein Hypnotikum; Bilignost wird als Röntgenkontrastmittel in Form einer Salzmischung in Lösung verwendet.
Literatur
1. Staatliches Arzneibuch der UdSSR / Gesundheitsministerium der UdSSR. - X-Hrsg. - M.: Medizin, 1968. - S. 78, 134, 141, 143, 186, 373,537
2. Staatliches Arzneibuch der UdSSR Bd. 1. Allgemeine Analysemethoden. Heilpflanzenrohstoffe / Gesundheitsministerium der UdSSR. - 11. Aufl., hinzufügen. - M.: Medizin, 1989. - S. 165-180, 194-199
3. Vorlesungsmaterial.
4. Pharmazeutische Chemie. In 2 Stunden: Lehrbuch / V. G. Belikov – 4. Aufl., überarbeitet. und zusätzlich - M.: MEDpress-inform, 2007. - S. 178-179, 329-332
5. Leitfaden für Laborkurse in pharmazeutischer Chemie. Herausgegeben von A.P. Arzamastseva, S. 152-156.
Gepostet auf Allbest.ru
Anhang 1
Arzneibuchartikel
Bilignost
Bis-(2,4,6-triiod-3-carboxyanilid)-adipinsäure
C 20 H 14 I 6 N 2 O 6 M. c. 1139,8
Beschreibung. Weißes oder fast weißes feinkristallines Pulver mit leicht bitterem Geschmack.
Löslichkeit. Praktisch unlöslich in Wasser, 95 % Alkohol, Ether und Chloroform, leicht löslich in Lösungen von Ätzalkalien und Ammoniak.
Authentizität. 0,001 % Lösung des Arzneimittels in 0,1 N. Natronlauge im Bereich von 220 bis 300 nm hat ein Absorptionsmaximum bei einer Wellenlänge von etwa 236 nm.
Beim Erhitzen von 0,1 g des Arzneimittels mit 1 ml konzentrierter Schwefelsäure werden violette Joddämpfe freigesetzt.
Farbe der Lösung. 2 g des Arzneimittels werden in 4 ml 1 N gelöst. Natriumhydroxidlösung, filtrieren und den Filter mit Wasser waschen, bis 10 ml Filtrat erhalten werden. Die Farbe der resultierenden Lösung sollte nicht intensiver sein als die Standardlösung Nr. 4b oder Nr. 4c.
Testen Sie mit Wasserstoffperoxid. Zu 1 ml der resultierenden Lösung 1 ml Wasserstoffperoxid hinzufügen; Innerhalb von 10-15 Minuten sollte keine Trübung auftreten.
Verbindungen mit einer offenen Aminogruppe. 1 g der Droge wird mit 10 ml Eisessig geschüttelt und filtriert. Zu 5 ml klarem Filtrat 3 Tropfen 0,1 molare Natriumnitritlösung hinzufügen. Nach 5 Minuten sollte die Farbe nicht intensiver sein als bei Standard Nr. 2g.
Säure. 0,2 g der Droge werden 1 Minute lang mit kochendem Wasser (je 4 mal 2 ml) geschüttelt und filtriert, bis ein klares Filtrat entsteht. Ich titriere die vereinigten Filtrate! 0,05 n. Natriumhydroxidlösung (Phenolphthalein-Indikator). Für die Titration sollten nicht mehr als 0,1 ml 0,05 N verwendet werden. Natronlauge.
Chloride. 2 g des Arzneimittels mit 20 ml Wasser schütteln und filtrieren, bis ein klares Filtrat entsteht. 5 ml Filtrat, mit Wasser auf 10 ml aufgefüllt, müssen den Chloridtest bestehen (nicht mehr als 0,004 % im Präparat).
Phosphor. 1 g der Droge wird in einen Tiegel gegeben und verascht, bis ein weißer Rückstand entsteht. Der Rückstand wird mit 5 ml verdünnter Salpetersäure versetzt und zur Trockne eingedampft, anschließend wird der Rückstand im Tiegel mit 2 ml heißem Wasser gut vermischt und durch einen kleinen Filter in ein Reagenzglas filtriert. Der Tiegel und der Filter werden mit 1 ml heißem Wasser gewaschen, das Filtrat im gleichen Reagenzglas gesammelt, dann 3 ml Ammoniummolybdatlösung hinzugefügt und 15 Minuten in einem Bad bei einer Temperatur von 38–40 ° belassen. Die Testlösung darf eine gelbliche Farbe haben, muss aber transparent bleiben (nicht mehr als 0,0001 % im Arzneimittel).
Jodmonochlorid. 0,2 g des Arzneimittels mit 20 ml Wasser schütteln und filtrieren, bis ein klares Filtrat entsteht. Zu 10 ml Filtrat 0,5 g Kaliumiodid, 2 ml Salzsäure und 1 ml Chloroform hinzufügen. Die Chloroformschicht sollte farblos bleiben.
Eisen. 0,5 g des Arzneimittels müssen den Eisentest bestehen (nicht mehr als 0,02 % im Arzneimittel). Der Vergleich erfolgt mit einem Standard, hergestellt aus 3,5 ml Standardlösung B und 6,5 ml Wasser.
Sulfatasche aus 1 g des Arzneimittels sollte 0,1 % nicht überschreiten.
Schwermetalle. Sulfatasche aus 0,5 g des Arzneimittels muss den Test auf Schwermetalle bestehen (nicht mehr als 0,001 % im Arzneimittel).
Arsen. 0,5 g der Droge müssen den Test auf Arsen bestehen (nicht mehr als 0,0001 % in der Droge).
Quantifizierung. Etwa 0,3 g der Droge (genau abgewogen) werden in einen 100-ml-Messkolben gegeben, in 5 ml Natronlauge gelöst, mit Wasser bis zur Marke aufgefüllt und gemischt. 10 ml der resultierenden Lösung werden in einen Kolben mit einem Fassungsvermögen von 250 ml gegeben, 5 ml einer 5 %igen Kaliumpermanganatlösung werden zugegeben und unter Rühren vorsichtig entlang der Kolbenwände 10 ml konzentrierte Schwefelsäure, 0,5, zugegeben Jeweils 1 ml wird zugegeben und 10 Minuten stehen gelassen. Dann langsam zugeben, 1 Tropfen nach 2-3 Sekunden, unter kräftigem Rühren. Natriumnitritlösung, bis sich die Flüssigkeit verfärbt und sich Mangandioxid auflöst. Danach sofort 10 ml einer 10 %igen Harnstofflösung zugeben und rühren, bis die Blasen vollständig verschwunden sind, dabei Natriumnitrit von den Kolbenwänden abwaschen. Anschließend werden der Lösung 100 ml Wasser, 10 ml einer frisch zubereiteten Kaliumjodidlösung zugesetzt und das freigesetzte Jod mit 0,1 N titriert. Natriumthiosulfatlösung (Indikator - Stärke).
1 ml 0,1 k. Natriumthiosulfatlösung entspricht 0,003166 g C 20 H 14 l 6 N 2 0 6, was in der Zubereitung mindestens 99,0 % betragen muss.
Lagerung. Liste B. In orangefarbenen Gläsern, vor Licht geschützt.
Röntgenkontrastmittel.
Jodoform
Triiodmethan
СНI 3 М.в. 393,73
Beschreibung. Kleine, lamellare, glänzende Kristalle oder feines kristallines Pulver von zitronengelber Farbe, scharfer, charakteristischer, anhaltender Geruch. Es ist bereits bei normalen Temperaturen flüchtig und wird mit Wasserdampf destilliert. Lösungen des Arzneimittels zersetzen sich unter dem Einfluss von Licht und Luft schnell und setzen Jod frei.
Löslichkeit. Praktisch unlöslich in Wasser, schwer löslich in Alkohol, löslich in Ether und Chloroform, schwer löslich in Glycerin. fettige und ätherische Öle.
Authentizität: 0,1 g des Arzneimittels werden in einem Reagenzglas auf einer Brennerflamme erhitzt; Dabei wird violetter Joddampf freigesetzt.
Schmelzpunkt 116–120° (mit Zersetzung).
Farbstoffe. 5 g der Droge werden mit 50 ml Wasser 1 Minute lang kräftig geschüttelt und filtriert. Das Filtrat sollte farblos sein.
Säure oder Alkalität. Zu 10 ml Filtrat 2 Tropfen Bromthymolblau-Lösung hinzufügen. Die erscheinende gelbgrüne Farbe sollte bei Zugabe von nicht mehr als 0,1 ml 0,1 N blau werden. Natronlauge oder Gelb durch Zugabe von nicht mehr als 0,05 ml 0,1 N. Salzsäurelösung.
Halogene. 5 ml des gleichen Filtrats, verdünnt mit Wasser auf 10 ml, müssen den Chloridtest bestehen (nicht mehr als 0,004 % im Präparat).
Sulfate. 10 ml des gleichen Filtrats müssen den Test auf Sulfate bestehen (nicht mehr als 0,01 % im Präparat).
Asche aus 0,5 g des Arzneimittels sollte 0,1 % nicht überschreiten.
Quantifizierung. Etwa 0,2 g des Arzneimittels (genau abgewogen) werden in einen Erlenmeyerkolben mit einem Fassungsvermögen von 250–300 ml gegeben, in 25 oder 95 % Alkohol gelöst und mit 25 ml 0,1 N versetzt. Silbernitratlösung, 10 ml Salpetersäure und 30 Minuten im Wasserbad unter Rückfluss kochen, dabei den Reaktionskolben vor Licht schützen. Der Kühlschrank wird mit Wasser gewaschen, 100 ml Wasser in den Kolben gegeben und das überschüssige Silbernitrat mit 0,1 N titriert. Ammoniumthiocyanatlösung (Indikator – Eisen-Ammonium-Alaun).
Gleichzeitig wird ein Kontrollexperiment durchgeführt.
1 ml 0,1 k. Die Silbernitratlösung entspricht 0,01312 g CHI 3, was in der Zubereitung mindestens 99,0 % betragen muss.
Lagerung. In einem gut verschlossenen Behälter, vor Licht geschützt, an einem kühlen Ort.
Gepostet auf Allbest.ru
Ähnliche Dokumente
Das Konzept der Brechung als Maß für die elektronische Polarisierbarkeit von Atomen, Molekülen, Ionen. Bestimmung des Brechungsindex zur Identifizierung organischer Verbindungen, Mineralien und Arzneimittel, ihrer chemischen Parameter sowie quantitativer und struktureller Analyse.
Kursarbeit, hinzugefügt am 05.06.2011
Grundlegende Abläufe bei der Arbeit in einem Labor für organische Chemie. Die wichtigsten physikalischen Konstanten. Methoden zur Bestimmung der Struktur organischer Verbindungen. Grundlagen der Struktur, Eigenschaften und Identifizierung organischer Verbindungen. Synthesen organischer Verbindungen.
Schulungshandbuch, hinzugefügt am 24.06.2015
Studium der theoretischen Grundlagen von Methoden zur Fällung organischer und anorganischer Arzneimittel. Analyse der Eigenschaften der Wechselwirkung von Arzneimitteln mit Indikatoren in Fällungsmethoden. Indikative Methoden zur Bestimmung des Titrationsendpunkts.
Kursarbeit, hinzugefügt am 30.01.2014
Oxidative Dimerisierung von Methan. Mechanismus der katalytischen Aktivierung von Methan. Herstellung organischer Verbindungen durch oxidative Methylierung. Oxidative Umwandlungen organischer Verbindungen mit einer Methylgruppe in Gegenwart eines Katalysators.
Dissertation, hinzugefügt am 11.10.2013
Berücksichtigung von Reaktionen, die auf der Bildung komplexer Metallverbindungen basieren und ohne deren Beteiligung. Das Konzept funktionalanalytischer und analytisch-aktiver Gruppen. Verwendung organischer Verbindungen als Indikatoren in titrimetrischen Methoden.
Kursarbeit, hinzugefügt am 01.04.2010
Chemische Struktur – die Reihenfolge der Verbindungen von Atomen in einem Molekül, die Reihenfolge ihrer Verbindung und gegenseitigen Beeinflussung. Die Verbindung von Atomen, aus denen organische Verbindungen bestehen; Abhängigkeit der Eigenschaften von Stoffen von der Art der Atome, ihrer Menge und der Reihenfolge des Wechsels.
Präsentation, hinzugefügt am 12.12.2010
Unter Isomerie versteht man das Phänomen der Existenz von Verbindungen mit identischer Zusammensetzung, aber unterschiedlicher Struktur und Eigenschaften. Interklassenisomerie wird durch die Art der funktionellen Gruppe bestimmt. Arten der räumlichen Isomerie. Arten der Nomenklatur organischer Verbindungen.
Präsentation, hinzugefügt am 12.03.2017
Grundlegende Methoden zur Vorhersage der Bildungsenthalpien organischer Verbindungen: Methoden der Molekularmechanik und additive Methoden. Benson-Methode und Tatevsky-Methode. Alkylbenzole und ihre funktionellen Derivate: Halogenbenzole, Polyphenyle, Pyridine.
Kursarbeit, hinzugefügt am 17.01.2009
Halogenierung aromatischer Verbindungen: Prozessmechanismus. Berechnung von Indikatoren für Mono- und Dichlorierung organischer Verbindungen. Reagenzverbrauch bei maximaler Ausbeute des Zielprodukts in komplexen Reaktionen. Auswahl eines geeigneten Reaktionsmechanismus.
Zusammenfassung, hinzugefügt am 15.02.2012
Leben als kontinuierlicher physikalischer und chemischer Prozess. Allgemeine Eigenschaften natürlicher Verbindungen. Klassifizierung von Naturstoffen mit niedrigem Molekulargewicht. Grundlegende Kriterien für die Klassifizierung organischer Verbindungen. Arten und Eigenschaften von Bindungen, gegenseitige Beeinflussung von Atomen.