Analyse fonctionnelle en laboratoire des composés organiques. Méthodes physicochimiques pour l'analyse des composés organiques
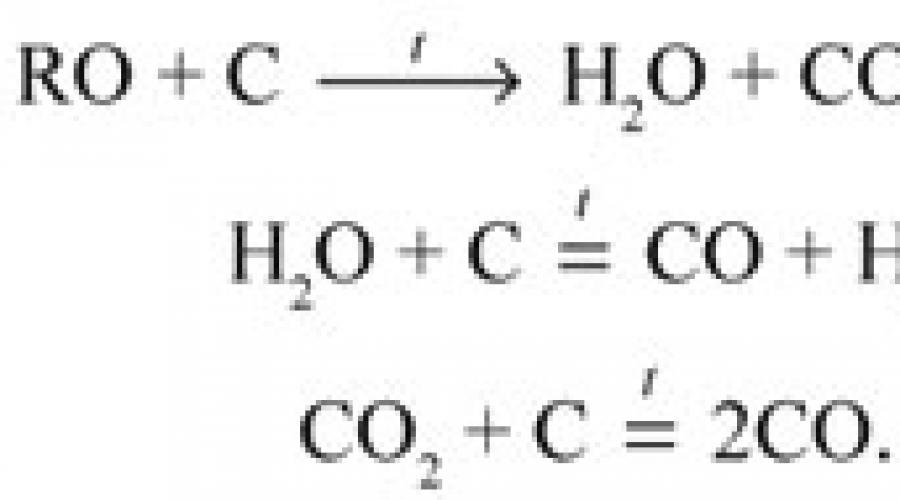
Lire aussi
Yu.V. Golubkov,
G.N. Golubkova
Détection de substances organiques
Pour les candidats aux universités, aux lycéens, lycées, gymnases, étudiants, ainsi qu'aux professeurs de chimie
Continuation. Voir le début dans le n°10/2010
Chapitre II.
Analyse élémentaire des substances organiques et détermination de leur structure
La matière organique purifiée est soumise à des analyses qualitatives et quantitatives. Le plus souvent, les composés organiques, en plus du carbone et de l'hydrogène, contiennent de l'oxygène, de l'azote, du soufre, des halogènes et du phosphore.
L'analyse qualitative et quantitative des composés organiques repose sur leur destruction (oxydation) suivie du dosage de CO 2, H 2 O, N 2, HCl, etc. par les méthodes conventionnelles.
L'analyse élémentaire des substances organiques a commencé avec la détermination du carbone et de l'hydrogène, il est donc intéressant d'examiner ce sujet plus en détail.
§ 7. Dosage analytique du carbone et de l'hydrogène
L'essence de la méthode.
Une quantité pesée du composé organique testé est oxydée avec l'un ou l'autre agent oxydant. Le monoxyde de carbone est obtenu quantitativement à partir du carbone et l'eau à partir de l'hydrogène. La masse de ces produits de combustion (oxydation) est déterminée en poids. Le tube de combustion (oxydation) est rempli de telle sorte que les autres éléments présents dans le composé organique n'interfèrent pas avec l'absorption quantitative finale du monoxyde de carbone (IV) et de l'eau dans des appareils d'absorption contenant des absorbeurs appropriés.
1) C + O 2 = CO 2,
le dioxyde de carbone est absorbé par l'eau de baryte (solution d'hydroxyde de baryum Ba(OH)2) et l'eau de chaux (solution d'hydroxyde de calcium Ca(OH)2), la chaux sodée (un mélange de 83 % de Ca(OH)2, 5 % de NaOH et 12 % H2O ) et d'autres substances.
2) H 2 + 1/2O 2 = H 2 O,
l'eau est absorbée par le perchlorate de magnésium anhydre Mg(ClO 4) 2 (anhydrone) ; en termes d'intensité de son effet desséchant, il se rapproche de l'oxyde de phosphore (V), ayant sur ce dernier l'avantage important que le sel de magnésium qui a absorbé l'eau et fondu par un chauffage prolongé à 220 ° C sous vide peut être à nouveau déshydraté et réutilisé. D'autres absorbeurs peuvent également être utilisés, par exemple le chlorure de calcium (de ce composé vient le nom de « tube de chlorure de calcium », voir ci-dessous).
Équipement.
Afin de déterminer si une substance donnée appartient à des composés organiques, il faut tout d'abord découvrir la présence de carbone dans celle-ci. Parfois, cela ne présente aucune difficulté : de nombreuses substances organiques se carbonisent lorsqu'elles sont chauffées, c'est-à-dire se transforment en charbon, ce qui indique la présence de carbone en eux. Mais dans certains cas, les substances contenant du carbone ne se carbonisent pas lorsqu'elles sont chauffées : par exemple, si vous chauffez de l'alcool, il s'évapore simplement, et s'il s'enflamme, il brûle sans suie. Par conséquent, le moyen le plus fiable de découvrir du carbone dans un composé organique consiste à brûler complètement ce composé et à détecter la présence de monoxyde de carbone (IV) dans les produits de combustion.
Afin de brûler la plus petite quantité possible de substance et de ne pas perdre les produits de combustion gazeux résultants, une petite quantité de matière organique est mélangée à un agent oxydant et chauffée. A cet effet, des agents oxydants trop actifs ne peuvent pas être utilisés, car la réaction qui se produit rendra difficile la capture des gaz émis par l'appareil. Si, par exemple, vous mélangez une substance combustible avec un agent oxydant aussi puissant que le chlorate de potassium KClO 3 (sel de Berthollet), alors, comme vous le savez, lorsque le mélange est chauffé, un éclair proche d'une explosion se produit. Par conséquent, dans ce cas, on utilise des agents oxydants peu actifs, qui n'agissent qu'avec une augmentation significative de la température, ce qui permet, en modifiant la température, de réguler le taux d'oxydation de la matière organique. Dans les combustions organiques, l'oxyde de cuivre (II) est généralement utilisé comme agent oxydant.
Pour définition qualitative prenez quelques milligrammes de la substance d'essai et mélangez-la avec une grande quantité (excès) d'oxyde de cuivre (II) granulaire, versez le mélange dans un tube à essai, fermez-le avec un bouchon avec un tube de sortie de gaz et chauffez-le. L'oxyde de cuivre (II) est réduit par la matière organique ; les produits d'oxydation de la matière organique sortant par le tube de sortie de gaz sont dirigés vers un récipient contenant une solution saturée d'hydroxyde de calcium (eau de chaux). Comme on le sait, la présence de monoxyde de carbone (IV) est détectée par la turbidité de l'eau de chaux. Si le combustible contient de l'hydrogène, l'eau formée par l'oxydation se dépose sous forme de gouttes de rosée sur les parties froides de l'appareil et peut ainsi être détectée.
Afin de produire quantification carbone et hydrogène, vous devez absorber les produits de combustion résultants avec quelque chose, chacun séparément, et les peser. Le principe de fonctionnement reste le même, seule la technique d'exécution change.
La méthode de combustion classique, développée autrefois par J. Liebig, est la suivante : la substance brûlée n'est pas mélangée à de l'oxyde de cuivre(II), mais est pesée dans une nacelle en porcelaine ou en platine, qui est ensuite placée dans un tube spécial pour combustion (Fig. 1). Il s'agit d'un long tube de verre réfractaire dans lequel est versée une longue couche d'oxyde de cuivre (II) granulaire, maintenue en place par deux petites spirales constituées d'un treillis de cuivre enroulé. Ces spirales pénètrent dans le tube avec un léger frottement et leur surface est pré-oxydée. Une spirale plus longue de treillis de cuivre oxydé est insérée dans l'extrémité opposée du tube de sorte qu'entre celui-ci et la couche d'oxyde de cuivre (II) il reste un espace plus long qu'un bateau en porcelaine avec une substance en suspension.
Le tube ainsi préparé est placé dans un four à combustion et, à l'aide de bouchons en caoutchouc dans lesquels sont insérés des tubes de verre, ses extrémités sont reliées à des dispositifs d'absorption pré-pesés : l'extrémité du tube dans laquelle se trouve la couche d'oxyde de cuivre(II) situé est relié à un tube de chlorure de calcium (Fig. 2), dans lequel la vapeur d'eau formée lors de la combustion de la substance brûlée est absorbée, et après lui un appareil à potassium rempli d'une solution concentrée d'hydroxyde de potassium et conçu pour absorber le monoxyde de carbone ( IV) est placé. En figue. La figure 3 montre les différentes conceptions des appareils Kali utilisés. L'extrémité opposée du tube de combustion est reliée à des gazomètres remplis : l'un d'air, l'autre d'oxygène (ils sont utilisés alternativement). Des dispositifs d'absorption sont également placés entre les gazomètres et le tube pour retenir l'humidité et le monoxyde de carbone (IV) pouvant être contenus dans les gaz remplissant les gazomètres.
Méthode de détermination.
Lorsque l'appareil est assemblé de cette manière, la partie du tube remplie d'oxyde de cuivre (II) est chauffée à une chaleur rouge foncé, puis la spirale de cuivre est retirée de la partie opposée du tube, un bateau avec la substance est introduit et, en poussant à nouveau la spirale, se ferme avec un bouchon, établissant la communication avec le gazomètre rempli d'air. Après cela, ils commencent à chauffer la longue spirale, puis augmentent soigneusement la température de la partie du tube dans laquelle se trouve le bateau contenant la substance, en faisant passer très lentement un courant d'air à travers le tube.
La substance présente dans le bateau s'évapore en partie et se décompose en partie, formant des substances volatiles. Ces vapeurs inflammables, se mélangeant à l'air, sont emportées par le courant de son jet jusqu'à la sortie du tube et, entrant en contact avec l'oxyde de cuivre(II) chaud, s'oxydent, réduisant l'oxyde de cuivre(II) en cuivre métallique. l'oxydation se produit trop rapidement et un trop grand nombre de ses produits (cela peut être jugé par le nombre de bulles traversant l'appareil) abaissent la température et réduisent l'intensité du flux d'air, sinon ils font le contraire ;
Si la substance brûlée s'évapore entièrement ou se décompose sans former de charbon, il ne restera plus rien dans le bateau à la fin de la combustion. Si du charbon se forme dans le bateau, cette partie du tube est chauffée plus fortement et, à la place de l'air, commence à passer de l'oxygène, dans lequel le charbon brûle progressivement.
Lorsque la combustion est terminée, l'air passe à nouveau à travers le tube jusqu'à ce que l'oxygène remplissant le tube et les dispositifs d'absorption soit déplacé par l'air. Cela doit être fait car les dispositifs d'absorption, lorsqu'ils ont été pesés avant de brûler, étaient remplis d'air et non d'oxygène. Après cela, le tube de chlorure de calcium et le pied à coulisse sont pesés, ce qui leur permet d'atteindre la température de la balance. Le gain de poids du tube de chlorure de calcium indique la quantité d'eau absorbée, et le gain de poids du tube de chlorure de calcium indique la quantité de monoxyde de carbone (IV) absorbée.
Liebig effectuait ses analyses à l'aide de charbon de bois : le tube de combustion était placé dans une auge et recouvert d'une couche de charbon brûlant. Pour abaisser la température en quelque endroit du tube, on ratissait le charbon ; pour augmenter la température, il était gonflé avec des soufflets fabriqués à la main. Avec l'introduction du gaz d'éclairage dans la pratique de laboratoire, des fours dotés d'un grand nombre de brûleurs à gaz ont commencé à être utilisés (Fig. 4), dont la flamme est facile à régler. Dans la pratique moderne, les fours électriques sont de plus en plus utilisés.
Méthode Liebig d'analyse organique, autrement appelée analyse élémentaire(détermination de la composition élémentaire de la matière organique) est encore largement utilisée en laboratoire. En plus de cette méthode, d’autres sont souvent utilisées. Par exemple, selon la méthode Denstedt, la combustion est réalisée sans oxyde de cuivre(II) ; la substance est brûlée dans un courant d'oxygène en présence de platine métallique, qui agit comme catalyseur. Ceci permet de réduire la taille du tube et de réduire le nombre de brûleurs dans le four.
Ces dernières années, la méthode de combustion simplifiée de G. Ter-Meulen et I. Gesling s'est de plus en plus répandue, qui consiste à brûler l'analyte dans un courant d'oxygène, en utilisant de l'oxyde de manganèse (IV) comme catalyseur ; Le four dans lequel s'effectue la combustion est d'une conception très simple et est chauffé par seulement deux brûleurs. Cette méthode est également pratique dans la mesure où elle permet d'utiliser de très petites quantités de substance pour la combustion et est méthode semi-micro.
Actuellement en utilisation microméthodes analyse organique. La conception spéciale des microbalances permet de peser de très petites quantités d'une substance avec une précision de 0,001 mg. Le tube de combustion et les dispositifs d'absorption ont été réduits et améliorés en conséquence.
La méthode originale de microanalyse organique a été développée par M.O. Korshun et V.A. Klimova. Son essence réside dans le fait que la combustion s'effectue dans un tube vide dans un courant d'oxygène. Un échantillon de 3 à 5 mg de la substance est placé dans un verre de quartz cylindrique, qui est inséré dans un tube de combustion (en position horizontale) ; Un courant d’oxygène traverse le tube. Lorsque la section du tube dans laquelle se trouve la coupelle contenant la substance combustible est chauffée, la décomposition et la carbonisation de la substance se produisent à l'intérieur de la coupelle, car l'oxygène ne peut pas circuler à l'intérieur de la tasse et n'est donc pas en excès. Les produits de décomposition thermique de la substance analysée, sortant de la coupelle dans un tube de combustion chauffé à 850-950°C, rencontrent un large excès d'oxygène et brûlent complètement. Le charbon restant dans la tasse brûle également. La combustion ne dure que 10 à 15 minutes. Ainsi, la microméthode de Korshun et Klimova est une méthode à grande vitesse.
S'il a été brûlé UN g de substance, prise de poids du tube de chlorure de calcium – R. g (masse d'eau formée), et le gain de poids du kaliapparat est R. 1 g (la masse du monoxyde de carbone formé (IV), le pourcentage d'hydrogène et de carbone dans la substance d'essai est déterminé comme suit. 1/9 de l'eau en masse est de l'hydrogène, donc dans R. g d'eau contient ( R./9) g d'hydrogène ; pour exprimer ce montant en pourcentage, il faut le multiplier par 100 et le diviser par le poids : R. 100/(9UN)%. Dans une molécule de monoxyde de carbone(IV), 12/44 de sa masse est constituée de carbone, cet échantillon contient donc (12 R. 1/44) g de carbone, soit 12 R. 1 100/(44UN) %.
Si la substance brûlée était constituée de carbone, d'hydrogène et d'oxygène, la masse de ce dernier est trouvée par la différence (en soustrayant de 100 % la somme des fractions massiques d'hydrogène et de carbone en pourcentage).
Lors de la détermination du carbone et de l'hydrogène à l'aide de la méthode Unterzaucher, l'analyte est brûlé sur de l'oxyde de cuivre (II) dans un courant d'air. L'hydrogène est déterminé par la quantité de H 2 O libéré, qui est lié au BaCl 2 anhydre, et le carbone est déterminé par la quantité de CO formée en faisant passer le CO 2 libéré sur du charbon chauffé à 1120°C. Dans ce cas, la teneur en CO est déterminée comme cela sera décrit ci-dessous (voir § 10). La méthode a été développée par J. Unterzaucher en 1950.
La littérature décrit de nombreuses autres options pour la conception instrumentale de méthodes de détermination du carbone et de l'hydrogène dans les composés organiques. Faisons attention à la rigueur des méthodes.
§ 8. Dosage analytique des halogènes
L'halogène dans les substances organiques n'est pas contenu sous forme d'ion, il ne peut donc pas être précipité avec du nitrate d'argent sous forme d'AgCl, AgBr ou AgI. Pour découvrir la présence d'un halogène dans une substance organique, il faut détruire cette dernière et ainsi libérer l'halogène ou le transformer en un autre composé dans lequel l'halogène est facile à ouvrir.
Un essai qualitatif pour l'halogène peut être effectué comme suit : la substance d'essai est collectée sur un fil de cuivre oxydé ou sur un morceau d'oxyde de cuivre (II) fixé dans l'œil d'un fil de cuivre et placé au feu ; la matière organique brûle, produisant de l'halogénure de cuivre, qui colore la flamme en vert ou vert bleuâtre. Cette méthode d'ouverture d'un halogène est connue sous le nom de Essais de Beilstein. De cette façon, vous pouvez découvrir la présence d'un halogène, mais vous ne pouvez pas décider lequel d'entre eux faisait partie de la substance étudiée. De plus, la réaction n'est pas sélective (les nitriles, certains dérivés pyridiniques... interfèrent).
Méthodes gravimétriques.
Pour déterminer quantitativement l'halogène dans les composés organiques, vous pouvez procéder comme suit : un échantillon de la substance à tester, un peu de nitrate d'argent et quelques millilitres d'acide nitrique concentré sont placés dans un tube en verre à paroi épaisse, scellé à une extrémité. Le tube est scellé et chauffé à 280-300 °C dans un four spécial, après avoir été préalablement placé dans un boîtier métallique, au cas où le tube éclaterait, incapable de résister à la pression. Dans ces conditions, la substance organique brûle (l'acide nitrique est un agent oxydant) et du chlorure, du bromure ou de l'iodure d'argent se forme dans le tube, selon l'halogène inclus dans la substance étudiée. Une haute pression se développe dans le tube en raison de la formation de monoxyde de carbone (IV) et d'oxydes d'azote. Une fois refroidi, le tube est ouvert, le précipité est éliminé, lavé, séché et pesé. Ce - méthode « combustion humide » de L. Carius(1860).
La détermination quantitative de l'halogène dans les dérivés halogènes solubles dans l'alcool a été considérablement simplifiée par A.V. Stepanov (1906). Le sodium métallique est ajouté morceau par morceau à une solution alcoolique d'une partie pesée du dérivé halogène et chauffé dans un ballon à reflux. Sous l'influence de l'hydrogène déplacé par le sodium de l'alcool, il se forme l'hydrocarbure et l'halogénure d'hydrogène correspondants qui, avec le sodium, donnent le sel correspondant, par exemple le chlorure de sodium. Ainsi, l'halogène passe à l'état ionique et est déterminé par des méthodes analytiques conventionnelles (gravimétriquement ou titrimétriquement).
Lorsque des substances contenant des halogènes sont brûlées pour déterminer le carbone et l'hydrogène, une spirale en maille d'argent est placée à l'extrémité du tube de combustion. Le but de la spirale est de retenir l'halogène libéré lors de la combustion de la substance, afin qu'il ne pénètre pas, avec le monoxyde de carbone (IV), dans le kalyapparat (fiole d'absorption) et ne soit pas absorbé par l'alcali.
Par méthode de « combustion à sec » la substance est brûlée dans un courant d'oxygène dans un tube de quartz d'un dispositif de combustion spécial, et les produits de décomposition passent sur des contacts en platine chauffés afin d'assurer une oxydation complète.
Par méthode de fusion « bombe » la substance organique contenant l'halogène est fusionnée dans une « bombe » avec un mélange de nitrate de potassium, de peroxyde de sodium et de sucre de canne (saccharose). Cela produit de l'halogénure de sodium, qui est précipité avec du nitrate d'argent et mis en suspension sous forme d'halogénure d'argent. La « bombe » utilisée pour la fusion (Fig. 5) est constituée d'un tube à essai métallique d'une profondeur de 25 mm et d'un diamètre interne de 13 mm ; il est constitué d'un alliage de chrome et de nickel. L'épaisseur des parois des tubes est de 1,5 mm ; il possède une bride de 3 mm de large sur laquelle sont placés le couvercle et le joint en plomb. Le fond rond du tube à essai est équipé d'une petite boucle. Le couvercle « bombe » est fermement fixé avec une pince en forme d’arc avec une vis bien ajustée.
Effectuer une analyse
Mélange. Peser 300 mg d'un mélange de nitrate de potassium et de sucre de canne (dans un rapport de 3:1) et 1,5 g de peroxyde de sodium dans un flacon peseur. Un tiers de ce mélange est chargé dans une « bombe » et la quantité appropriée d’analyte est ajoutée. Ensuite, le reste du mélange est ajouté, le couvercle de la « bombe » est bien vissé et le contenu est soigneusement mélangé en secouant. Ce mélange minutieux est très important pour obtenir une oxydation complète. Par des tapotements répétés, le contenu de la « bombe » est forcé de s'accumuler au fond - le mélange réactionnel est prêt à fusionner.
Fusion La « bombe » est soutenue par la tête d'une pince à vis avec une pince ou un morceau de fil solide inséré dans le trou de la tête de la vis, et est progressivement descendue d'un tiers dans la partie supérieure de la petite flamme incolore. d'un brûleur à gaz (il faut éviter de chauffer les « bombes » trop près de son couvercle). La fusion se termine dans 10 s ; la fusion elle-même peut être facilement reconnue par le tremblement de la « bombe ». Théoriquement, la fusion peut être considérée comme complète après les 10 s spécifiées, mais il est recommandé de maintenir la « bombe » dans la flamme pendant 5 s supplémentaires pour être sûr de la fusion complète de l'ensemble du mélange réactionnel. Ensuite, la « bombe » est refroidie avec de l’eau sous le robinet.
Refroidissement et filtrage. Tout d'abord, l'extérieur de la « bombe » est lavé avec de l'eau distillée. Ensuite, il est ouvert et l'intérieur du couvercle est lavé avec de l'eau distillée chaude ; L'eau de lavage est collectée dans un tube de sédimentation. Ensuite, le tube « bombe » est inséré dans le tube de sédimentation et de l'eau distillée chaude est à nouveau ajoutée afin qu'elle recouvre complètement le tube « bombe ». Pour dissoudre complètement le mélange réactionnel, secouez-le. Une fois tout le mélange dissous, le tube à essai est soulevé avec un fil de platine épais et soigneusement lavé avec de l'eau distillée chaude. Ensuite, le tube à essai est pris par l'anse avec une pince à pointes en platine et lavé à l'intérieur avec de l'eau distillée chaude, qui est versée dans la solution originale.
DÉTERMINATION DU CHLORE. La solution est refroidie dans un bain de glace et acidifiée avec de l'acide nitrique concentré. La solution acidifiée est filtrée dans un autre tube à essai pour précipitation ; 1 ml d'une solution de nitrate d'argent à 5 % est ajouté au filtrat, chauffé au bain-marie et le précipité est filtré.
Le précipité de chlorure d'argent est séché jusqu'à poids constant et pesé, après quoi la teneur en chlore de la substance analysée est calculée.
Détermination du brome et de l'iode. La solution principale et les eaux de lavage sont neutralisées en présence de phénolphtaléine avec de l'acide nitrique jusqu'à ce qu'elles deviennent légèrement roses. 100 mg de sulfate d'hydrazine sont ajoutés et le mélange est chauffé au bain de vapeur pendant 15 minutes. Après avoir filtré ce mélange dans un autre tube à essai de précipitation, il est acidifié avec 0,5 ml d'acide nitrique concentré, 1 ml d'une solution à 5% de nitrate d'argent est ajouté, le tout est à nouveau chauffé au bain de vapeur et le précipité est filtré, séché et pesé (comme pour la détermination du chlore).
Méthodes titrimétriques.
Détermination du chlore et du brome. Une substance organique contenant du chlore et du brome est ajoutée au ballon de réaction et décomposée à 115-125 °C avec de l'acide sulfurique concentré en présence d'un mélange de bichromate de potassium et de bichromate d'argent. La combustion s'effectue sous courant d'oxygène. L'halogène (X) entre dans un récipient contenant un excès de solution de soude de concentration molaire de 0,01 mol/l, auquel est ajouté 1 ml de peroxyde d'hydrogène :
2RX -> X2 + X CO2+ X H 2 O (oxydation),
X 2 + 2NaOH + H 2 O 2 = 2NaX + O 2 + 2H 2 O (absorption).
Une solution d'hydroxyde de sodium en excès est titrée avec une solution acide de même concentration et le pourcentage de chlore ou de brome est calculé à partir de la quantité d'alcali consommée. La présence d'iode n'interfère pas avec la détermination, puisqu'il est oxydé en iodate. Cette méthode ne convient pas aux substances volatiles ou aux substances contenant également de l'azote ou du soufre.
Détermination de l'iode. La matière organique contenant de l'iode est brûlée en présence d'un catalyseur dans une atmosphère d'oxygène ; cela produit de l'iode, du monoxyde de carbone (IV) et de l'eau. L'iode est capturé et oxydé avec du brome pour former de l'acide iodique HIO 3, ce dernier est réduit avec de l'iodure d'hydrogène en iode, qui est déterminé par iodométrie.
Le processus se déroule selon les équations suivantes :
2RI -> Je 2 + X CO2+ X H 2 O (combustion),
HIO 3 + 5HI = 3I 2 + 3H 2 O,
I 2 + 2Na 2 S 2 O 3 = 2NaI + Na 2 S 4 O 6 (titrage iodométrique).
§ 9. Dosage analytique de l'azote
Détermination qualitative de l'azote. Dans les produits de combustion de substances organiques contenant de l'azote, ce dernier est à l'état libre. Par conséquent, afin de déterminer la présence d’azote dans un composé organique, il est nécessaire de le détruire et de convertir l’azote en un composé qui peut être facilement découvert par certaines réactions qualitatives.
Par exemple, selon la méthode Lassen l'azote est détecté de cette manière : dans un tube de verre scellé à une extrémité, un peu de la substance étudiée est calciné avec un morceau de potassium métallique ; dans ce cas, la substance est carbonisée et le cyanure de potassium se forme à partir du potassium, du carbone et de l'azote contenus dans la matière organique étudiée. Le tube est immergé dans une petite quantité d'eau, ce qui entraîne son éclatement et le cyanure de potassium se dissout dans l'eau. La solution est filtrée des fragments de verre et du charbon. À la solution résultante contenant du cyanure de potassium, ajoutez des solutions de sels de fer (II), par exemple du sulfate de fer (II), et des sels d'oxyde de fer (III), par exemple du chlorure de fer (III). L'hydroxyde de potassium contenu dans la solution précipite les hydroxydes de fer (II) et (III), et lorsque le cyanure de potassium réagit avec les sels de fer (II), de l'hexacyanoferrate de potassium (II) K4 se forme :
6KCN + FeSO 4 K 4 + K 2 SO 4.
Après cela, de l'acide chlorhydrique (chlorhydrique) est ajouté, ce qui entraîne la formation de sels de fer. Après dissolution des hydroxydes de fer(II) et (III), un précipité de bleu de Prusse formé à partir d'hexacyanoferrate de potassium(II) et de chlorure de fer(III) (reste de l'hexacyanoferrate de fer(III) Fe 4 3) :
3K 4 + 4FeCl 3 = Fe 4 3 + 12KCl.
La formation de bleu de Prusse (un sel insoluble dans l'eau avec une couleur bleue caractéristique) indique la teneur en azote de la substance étudiée.
Dosage quantitatif de l'azote (méthode Dumas). La matière organique mélangée à l'oxyde de cuivre (II) est brûlée en faisant passer un flux de dioxyde de carbone à travers un tube de combustion ; les produits de combustion sont collectés dans un appareil rempli d'une solution concentrée d'hydroxyde de potassium. Le dioxyde de carbone est absorbé, le volume d'azote libre restant est mesuré puis sa masse est calculée.
Lorsque des substances organiques contenant de l'azote sont brûlées, une spirale de cuivre réduite est placée à l'extrémité du tube de combustion : si une petite quantité d'oxydes d'azote se forme, ils, au contact du cuivre chaud, lui cèdent de l'oxygène, et l'azote est obtenu dans un état libre.
L'azote dégagé est collecté quantitativement dans un compteur d'azote (récipient gradué) sur une solution de potasse à 50 %. La méthode a été développée par J.B.A. Dumas en 1831.
La détermination gazométrique de l'azote par la méthode Dumas est applicable aux composés organiques contenant de l'azote sous toute forme, à savoir : les composés amino-, nitroso-, azo-, cyano, les nitrites d'alkyle, ainsi que les nitrates et les composés hétérocycliques contenant de l'azote.
Lors de la détermination de l'azote dans les composés organiques (principalement les amines) à l'aide de la méthode Kjeldahl, l'analyte est décomposé avec du H 2 SO 4 concentré en présence d'un catalyseur, généralement du mercure ou de ses sels. Dans ce cas, après alcalinisation, l'ammoniac est isolé, qui est distillé, absorbé avec une solution de H 2 SO 4 ou HCl et déterminé titrimétriquement ou colorimétriquement avec le réactif de Nessler - une solution alcaline de tétraiodomercurate de potassium (II) dihydraté K 2 [HgI 4 ] 2H 2 O. Parfois, pour obtenir des résultats plus précis, la substance contenant de l'azote est prétraitée avec un agent réducteur, par exemple HI. La méthode a été développée par I. Kjeldahl en 1883.
Il existe des dispositifs d'analyse semi-automatiques qui permettent de déterminer la teneur en azote simultanément avec la teneur en carbone et en hydrogène.
§ 10. Détermination analytique de l'oxygène
L'une des premières méthodes de détermination de l'oxygène dans les composés organiques est la réduction d'un flux d'hydrogène (hydrogénation) à une température de 1 120 °C. Cela produit de l'eau.
De plus, les composés organiques peuvent être détruits par fusion avec du potassium ou du sodium métallique. Dans ce cas, l’oxygène forme un oxyde métallique facile à détecter. Cependant, si le composé contient simultanément de l’oxygène et de l’azote, alors un cyanate de métal alcalin, le sel de l’acide cyanique HNCO, peut se former.
Selon la méthode Unterzaucher, un composé organique contenant de l'oxygène est soumis à une pyrolyse dans une atmosphère d'azote. Les produits gazeux passent sur du charbon chauffé à 1 110 - 1 150 °C. Dans ce cas, il se forme de l'oxyde de carbone (II), qui réagit ensuite avec l'oxyde d'iode solide (V) (anhydride iodique) I 2 O 5 :
5CO + I 2 O 5 = 5CO 2 + I 2.
L'iode libéré est dosé par titrimétrie à l'aide de thiosulfate de sodium Na 2 S 2 O 3 :
2Na 2 S 2 O 3 + I 2 = 2NaI + Na 2 S 4 O 6.
Cette méthode a été développée par J. Unterzaucher en 1940.
Certains chercheurs préfèrent utiliser un tube de quartz vertical en forme de T rempli de carbone (Figure 6).
Microdétermination directe de l'oxygène dans les composés organiques.
La substance est soumise à une décomposition réductrice sur du charbon dans une atmosphère inerte. Dans ce cas, tout l'oxygène se transforme en monoxyde de carbone (II) :

![]()
M.O. Korshun a apporté une grande contribution au développement de la méthode de microdétermination de l'oxygène et d'autres éléments. Elle a proposé une détermination gravimétrique de l'oxygène basée sur trois facteurs :
1) par la perte de masse de I 2 O 5 (voir l'équation précédente) ;
2) par gain de poids de cuivre :
Cu + I = CuI 2 ;
3) par prise de poids en ascarite* :
CO 2 + 2NaOH = Na 2 CO 3 + H 2 O.
§ 11. Dosage analytique du soufre
La matière organique contenant du soufre est oxydée selon la méthode Carius « combustion humide » dans de l'acide nitrique concentré dans un tube de verre scellé à paroi épaisse en présence de chlorure de baryum à une température de 280 à 300 °C. Le soufre présent dans la substance est oxydé en acide sulfurique, qui réagit ensuite avec le chlorure de baryum pour former du sulfate de baryum :
H 2 SO 4 + BaCl 2 = BaSO 4 + 2HCl.
Après refroidissement, le tube est ouvert, le précipité est éliminé, lavé, séché et pesé (analyse gravimétrique).
Par méthode de « combustion à sec » une substance organique contenant du soufre, mais ne contenant pas d'halogènes ni d'azote, est oxydée dans une atmosphère d'oxygène en présence d'un catalyseur au platine. Dans ce cas, le soufre est transformé en oxydes gazeux, absorbés par une solution aqueuse de peroxyde d'hydrogène :
SO 2 + SO 3 + H 2 O 2 + H 2 O = 2H 2 SO 4.
Ainsi, le produit final de l'oxydation du soufre est l'acide sulfurique, qui est titré avec une solution de soude de concentration molaire de 0,01 mol/l (analyse titrimétrique) :
H 2 SO 4 + 2NaOH = Na 2 SO 4 + 2H 2 O.
§ 12. Dosage analytique du phosphore et de l'arsenic
Pour doser le phosphore, la matière organique est décomposée soit par fusion avec des mélanges oxydants solides, soit par traitement avec un mélange de peroxyde d'hydrogène et d'acide sulfurique concentré. Dans ce cas, le phosphore présent dans la matière organique est transformé en acide orthophosphorique, qui est ensuite dosé en poids (analyse gravimétrique) sous forme de phosphoromolybdate d'ammonium, préalablement séché à l'éther.
Pour déterminer l'arsenic, la matière organique est oxydée de la même manière que les composés contenant du phosphore. Dans ce cas, ils utilisent soit la « combustion humide » (méthode Carius), soit la fusion dans une « bombe ». L'acide arsénique H 3 AsO 4 obtenu est dosé par gravimétrie (en poids) sous forme de sel de magnésium de l'acide pyromésique :
AsO 3- 4 + Mg 2+ + NH 4 + = MgNH 4 AsO 4,
2MgNH 4 AsO 4 Mg 2 As 2 O 7 + H 2 O + 2NH 3;
ou par voie iodométrique (méthode titrimétrique ou volumétrique) :
H 3 AsO 4 + 2HI = H 3 AsO 3 + I 2 + H 2 O,
Je 2 + 2Na 2 S 2 O 3 = 2NaI + Na 2 S 4 O 6.
§ 13. Détermination de la structure des substances organiques
La chimie est un domaine de connaissances scientifiques et d'activités pratiques qui se développe grâce à la synthèse de nouveaux composés et à la création de matériaux à partir de ceux-ci. La tâche principale d'un chercheur en chimie après la synthèse d'un composé est d'établir la structure de la molécule.
Déterminer la structure complète d’une molécule, y compris géométrique et électronique, est une tâche complexe qui ne peut être complètement résolue. Par conséquent, les détails de la structure moléculaire d'un composé plus ou moins complexe peuvent être étudiés à l'infini, en y ajoutant de plus en plus d'informations nouvelles.
Considérons les fondements physiques des méthodes les plus importantes pour déterminer la structure des molécules de composés organiques. Ces méthodes sont appelées méthodes de recherche physique. Les méthodes chimiques permettant de prouver la structure géométrique d'une molécule (structure géométrique) n'ont pas non plus perdu de leur importance.
Au moment de l'émergence de la théorie structurale d'A.M. Butlerov, une vérité importante a été réalisée : la structure d'une molécule détermine toutes ses propriétés chimiques, et de la somme des propriétés chimiques, on peut tirer une conclusion correcte sur la structure d'un substance. Cette vérité n’a en aucun cas perdu son sens à l’ère des méthodes de recherche physique. L'analyse et la synthèse restent constantes en chimie ; Ces concepts constituent, on le sait, la base de la pensée, la base de toute connaissance. Synthétisant des substances de plus en plus complexes, le chimiste connaît à l'avance les fragments initiaux à partir desquels se forme la structure du nouveau composé. Il assume donc une structure à l’avance. Après la synthèse, il ne reste plus qu'à le prouver. Si la structure d'un composé naturel sur lequel il n'existe aucune information est déterminée, ses propriétés chimiques sont d'abord étudiées, ce qui permet de tirer des conclusions sur la présence de certains éléments structurels. L'étape la plus importante ici est l'analyse chimique - la décomposition d'une molécule complexe en fragments, en « éléments constitutifs » individuels et l'établissement de la nature de leur combinaison dans la molécule.
Au cours des dernières décennies, l'analyse chimique a été complétée analyse spectrale de masse– une méthode physique basée sur la mesure de la masse de particules de matière chargées formées lorsque des molécules complexes sont bombardées par un puissant flux d’électrons. Cette méthode est traditionnellement toujours considérée avec les méthodes de spectroscopie optique. Les méthodes mentionnées ci-dessus permettent d'établir la présence d'éléments individuels essentiels de la géométrie (spectroscopie IR, spectroscopie Raman) et électronique (spectroscopie électronique, spectroscopie de résonance magnétique nucléaire, spectroscopie de résonance paramagnétique électronique, spectroscopie électronique des rayons X, etc. ) structure de la molécule et de mieux la comprendre pleinement.
Spectroscopie de résonance magnétique nucléaire (spectroscopie RMN)– l’une des méthodes les plus modernes pour étudier la structure des composés organiques. Outre la spectroscopie électronique et vibrationnelle, la méthode RMN, apparentée à la spectroscopie de résonance, permet de résoudre les questions les plus subtiles de la chimie structurale, notamment celles de l'état électronique des atomes dans une molécule. La méthode convient à l’étude de molécules contenant des atomes avec un nombre impair de protons ou de neutrons. Les noyaux de ces atomes ont un moment magnétique et sont paramagnétiques.
Les fondements théoriques de ces méthodes d’étude de la structure des composés organiques sont complexes et sont abordés dans les cours pertinents de l’enseignement supérieur.
* L'ascarite est de l'amiante imprégnée de NaOH fondu. – Note éd.
Les bases de la définition ont été développées par F. Pregl. Un échantillon de 3 à 5 mg de la substance est brûlé à une température de 900 °C dans un courant d'oxygène purifié de l'hydrogène, de l'eau et du dioxyde de carbone. L'hydrogène est purifié de l'oxygène en faisant passer le gaz sur un catalyseur en platine à 800 °C. L'élimination complète du dioxyde de carbone et de l'eau est réalisée par passage au perchlorate de magnésium anhydre ( anhydre) et à travers de l'amiante imprégnée de soude caustique fondue ( ascarite).
Après le tube de combustion, des tubes avec absorbeurs sont installés : anhydrone et ascarite. Le gain de poids du premier absorbeur correspond à la quantité d'eau à partir de laquelle est calculée la teneur en hydrogène d'un échantillon de la substance ; le gain de poids du deuxième absorbeur donne la quantité de dioxyde de carbone, qui est utilisée pour déterminer la teneur en carbone de la substance analysée.
Les halogènes et le soufre peuvent être déterminés en décomposant la substance selon Carius. Halogènes sous forme d'halogénure d'argent - par gravitation ou par titrage de l'excès de nitrate d'argent. Le soufre est dosé sous forme de sulfate de baryum. Après la combustion de l’échantillon, les gaz passent sur une couche de cuivre métallique, où les oxydes d’azote sont réduits en azote libre. L'azote est déterminé par la méthode volumétrique par la quantité de gaz non absorbée.
Détermination du poids moléculaire
Pour déterminer le poids moléculaire d'un composé, des méthodes de cryoscopie basées sur la loi de Raoult sont souvent utilisées. Pour ce faire, déterminez le point de congélation du solvant puis de la solution. La différence est directement proportionnelle au nombre de molécules de la substance dissoutes dans une masse donnée de solvant. La masse moléculaire est déterminée par la formule :
Où R.- le poids de la substance ; P - poids du solvant ; K - constante cryoscopique ;
De même, dans la méthode ébullioscopique, le poids moléculaire est déterminé par la différence entre les points d’ébullition d’un solvant pur et d’une solution.
Pour les composés de poids moléculaire élevé, les méthodes décrites ci-dessus sont totalement inapplicables. Dans ce cas, trois méthodes sont utilisées : viscométrique, osmotique et sédimentation, qui, à leur tour, ne sont pas applicables aux substances de poids moléculaire normal.
Actuellement, la spectrométrie de masse est le plus souvent utilisée pour déterminer la masse moléculaire d'une substance inconnue.
Méthodes d'isolement de substances individuelles
Pour isoler les composés, les méthodes physiques suivantes sont utilisées : différents types de distillation - fractionnée à pression atmosphérique, sous vide, sous vide poussé, distillation moléculaire, cristallisation, extraction, chromatographie. De plus, il existe de nombreuses méthodes spéciales qui prennent en compte les spécificités du groupe fonctionnel.
· Distillation moléculaire . Pour les substances qui se décomposent au point d’ébullition même sous vide poussé, la « distillation moléculaire » est utilisée. Son principe est que sous un vide poussé (10 -5 -10 -8 mm Hg) à partir de la surface chauffée de la substance fondue à distiller, les molécules passent dans la phase gazeuse à une température bien inférieure au point d'ébullition du composé donné. . La vapeur de la substance se condense alors sur la surface froide. Cela permet de purifier des substances ayant un poids moléculaire relativement important et une structure fragile.
· Distillation à la vapeur . Comme on le sait, une substance bout à une température où sa pression de vapeur est égale à la pression atmosphérique. Si vous chauffez deux liquides non miscibles, ils bouilliront à une température où la pression de vapeur totale des deux liquides est égale à la pression atmosphérique. L'eau est utilisée comme deuxième liquide. Ainsi, la distillation de ce mélange de liquides peut être effectuée en dessous de 100°C. La quantité des deux substances dans le distillat est déterminée par le rapport entre le produit de la pression de vapeur de chaque substance et son poids moléculaire.
· Cristallisation . La recristallisation est utilisée pour purifier les solides. La méthode est basée sur le fait que pour la plupart des composés, lorsque leurs solutions sont refroidies, la solubilité de la substance diminue.
· Extraction . Méthode de séparation basée sur la différence des coefficients de répartition d'une substance entre deux liquides non miscibles.
Chromatographie
Ø Chromatographie – méthode de séparation basée sur différentes vitesses de déplacement des zones de concentration des composants du mélange étudié dans l'écoulement de la phase mobile ( éluant) par rapport à la phase stationnaire.
§ Par tâches à résoudre allouer préparatoire(séparation quantitative des substances) et analytique chromatographie (détection de substances et caractéristiques quantitatives et qualitatives des mélanges).
§ Selon les principes de séparation la chromatographie est divisée en adsorption, distribution, échange d'ion Et tamis.
v Chromatographie d'adsorption . Les substances à séparer doivent différer par leur affinité pour l'adsorbant solide, qui est la phase stationnaire. L'oxyde d'aluminium et le gel de silice sont généralement utilisés comme adsorbants. Le charbon actif, le sulfate de baryum, le silicate de magnésium et les polyamides sont beaucoup moins fréquemment utilisés.
La capacité des substances à être adsorbées sur un adsorbant polaire est largement déterminée par leur polarité. En fonction de leur capacité à être adsorbées, les substances possédant différents groupes fonctionnels peuvent être classées dans l’ordre suivant :
RH< ROCH 3 < R-NO 2 < R-N(CH 3) 2 < R-COOCH 3 < R-NH 2 < R-OH < R-CONH 2 < R-COOH.
En termes de polarité, et donc en termes de capacité d'élution, les solvants éluants forment la série suivante :
H 2 O > CH 3 OH > C 2 H 5 OH > CH 3 COCH 3 > CH 3 COOC 2 H 5 > C 2 H 5 OC 2 H 5 > CHCl 3 > CCl 4 > cyclohexane > n-hexane
L'élution s'effectue soit avec un éluant (mélange d'éluants), soit successivement avec plusieurs éluants en passant du moins polaire au plus polaire, soit avec un mélange de deux solvants (en augmentant successivement la concentration du plus polaire).
· Principales variantes de chromatographie d'adsorption .
§ Chromatographie d'adsorption sur colonne. L'adsorbant est placé dans une colonne. La substance à séparer est d'abord appliquée sur le dessus, puis passe l'éluant qui se déplace sous l'influence de la gravité ou est pompé sous pression par une pompe spéciale.
La séparation des substances est contrôlée soit par des méthodes physico-chimiques (détection UV, réfractométrie) soit par des méthodes analytiques chromatographiques.
§ Chromatographie sur couche mince (CCM). Le sorbant est déposé en couche mince sur un substrat en verre, en aluminium ou en plastique.
La couche absorbante peut être lâche ou fixée à l'aide de produits chimiques spéciaux (amidon, gypse). Un échantillon de la substance est déposé au fond de la plaque, qui est ensuite placée dans une boîte contenant un éluant. Le solvant monte le long de la plaque sous l’effet des forces capillaires ( Ascendant chromatographie), produisant une séparation. Dans le cas de substances difficiles à séparer, recourir à bidimensionnel chromatographie, lorsque la substance est d'abord éluée dans une direction, puis l'élution est effectuée dans une direction perpendiculaire.
Dans les conditions modernes, des plaques fabriquées industriellement pour une CCM préparative ou analytique sont généralement utilisées.
· Identification et caractérisation des substances. Les composés colorés sont observés directement lors de la chromatographie. Les substances incolores doivent être « identifiées » – converties en composés colorés.
En fonction du sorbant, du fixateur et de la nature de la substance séparée, diverses méthodes de « détection » sont utilisées, par exemple, les glucides sont carbonisés à haute température, y compris après pulvérisation de solutions d'acide sulfurique, les acides aminés donnent des produits colorés après traitement avec un solution de ninhydrine. Sur la base de l'intensité de la couleur des composés séparés avec des réactifs spécifiques, leur teneur dans le mélange est jugée.
Pour caractériser les substances, le terme « mobilité chromatographique", qui est noté Rf, est le rapport entre la plage de la zone de substance et la plage de l'éluant.
v Chromatographie de partage . Cette version de la chromatographie est basée sur la répartition des substances entre la phase mobile (gaz, liquide) et la phase stationnaire - liquide maintenue sur un support solide inerte. Les plus utilisées sont la chromatographie de partage sur papier et la chromatographie gaz-liquide.
§ Chromatographie sur papier. La base de la chromatographie sur papier est la répartition d'un mélange de substances séparées entre de l'eau adsorbée sur le papier et un solvant saturé d'eau. Grâce à cette méthode, la séparation et l’identification des acides aminés et des monosaccharides ont été réalisées avec succès. Actuellement, cette option de chromatographie a perdu de sa pertinence.
§ Chromatographie gaz-liquide. Il s'agit d'une chromatographie de partage entre une phase liquide stationnaire supportée par un support et un gaz (généralement de l'hélium, de l'azote ou de l'hydrogène).
Pour caractériser les substances séparées, utilisez « temps de rétention" Il s'agit du temps écoulé entre le moment où le mélange est introduit dans la colonne et celui jusqu'à ce qu'il sorte de la colonne et fasse passer la substance à travers un détecteur approprié, par exemple un détecteur qui enregistre les changements de conductivité thermique. Cette option est l’une des méthodes chromatographiques les plus utilisées, notamment à des fins analytiques.
v Chromatographie d'échange d'ions . La méthode est basée sur la répartition des substances chargées (ions) entre les phases mobile et stationnaire en fonction de leur affinité pour les centres ioniques de la phase stationnaire.
Selon la nature de l'échangeur d'ions, il existe cationique Et anionique chromatographie. L'eau, les solutions d'acides et d'alcalis et les solutions tampons sont largement utilisées comme éluants. Les matériaux échangeurs d'ions les plus courants sont les échangeurs de cations et les échangeurs d'anions à base de polymères réticulés contenant des groupes fonctionnels ionogènes, ainsi que de cellulose modifiée.
v Chromatographie sur tamis (chromatographie sur gel) . Une caractéristique distinctive de la chromatographie sur gel est que dans les gels formés de macromolécules tridimensionnelles « réticulées », il existe des pores de certaines tailles, dans lesquels pénètrent les plus petites molécules des molécules séparées et n'entrent pas dans les plus grosses. Par conséquent, contrairement à la chromatographie par adsorption, en chromatographie sur gel, les plus grosses molécules traversent la colonne en premier et les petites en dernier. La colonne de séparation est remplie de grains de gel lyophile ou hydrophile. Des exemples de tels matériaux chromatographiques sont les gélifiants naturels modifiés - agar, dextrines, séphadex (dextranes réticulés) et les tamis synthétiques à base de polyacrylamide ou de polystyrène « réticulé ».
Berzelius J. (1779 1884) – chimiste suédois. La recherche scientifique couvre tous les principaux problèmes de la chimie de la première moitié du XIXe siècle.
Wehler F. (1800-1882) – chimiste allemand. Travaille en chimie inorganique et organique. Avec J. Liebig, il établit l'isomérie des sels d'acide explosif.
3 Gmelin L. (1788-1853) – chimiste allemand. Il a publié des ouvrages de référence sur les données expérimentales, qui ont connu plusieurs éditions.
Liebig J. (1803-1873) – chimiste allemand. Créateur de la théorie des radicaux, fondateur de l'agrochimie. Acides organiques étudiés.
Butlerov A. (1828-1886) - Chimiste russe, créateur de la théorie de la structure des composés organiques. Isomérie prévue de nombreux composés.
Gérard S. (1816-1856) – chimiste français. Il a travaillé pour Yu. Liebig, a écouté les conférences de J. Dumas. Création d'une théorie des types. De nombreux chimistes russes ont étudié avec C. Gerard.
Schorlemmer K. (1834 – 1892) – chimiste organique allemand. Il a travaillé dans le domaine des alcanes et a des ouvrages sur l'histoire de la chimie.
Lossen V. (1838 - 1905) - chimiste allemand. Les principaux travaux sont liés à l'étude des alcaloïdes, à la découverte du réarrangement des acides hydroxamiques.
Carius L. (1829-1875) – chimiste allemand. Développé une méthode pour déterminer le soufre, les halogènes et d'autres éléments dans les composés organiques (1860)
Beilstein F. (1838-1906) – chimiste organique russe. Principaux travaux dans le domaine de la synthèse des composés aromatiques. Initiateur et premier compilateur de l'ouvrage de référence en plusieurs volumes sur les composés organiques (Handbuch der organische Chemie), connu sous le nom de Belstein Handbook.
Pregl F. (1869-1930) – chimiste allemand. Fondateur de la microanalyse de composés organiques. Prix Nobel 1923
Raoul F. (1830-1901) – chimiste français. La direction principale de la recherche est l'étude des solutions.
ANALYSE DES SUBSTANCES ORGANIQUES
(analyse organisationnelle dépassée), qualités. et les quantités. détermination de la composition de l'org. in-in et établissement de leur structure.
Lors de la détermination des qualités. composition de l'org. in-in utilise une variété de méthodes basées sur la chimie. p-tions, accompagnées de la formation de produits aux qualités caractéristiques (couleur, odeur, température de fusion, etc.), et sur des mesures physiques. et physico-chimique (chromatographiques, spectrales, etc.) des composés identifiés.
Avec quantités, analyse de l'org. la quantité de réactif entrée dans la distribution auprès de l'organisation déterminée est établie. conn., ou mesurer diff. physique et physico-chimique caractéristiques associées au nombre de composés déterminés.
O.v. UN. comprend analyse élémentaire, groupe structurel (y compris fonctionnel et stéréospécifique), analyse moléculaire, analyse de phase Et analyse structurelle.
Historiquement, les méthodes d'analyse élémentaire de l'organisation ont été les premières à être développées. in-in (A. Lavoisier, fin XVIIIe siècle), basé sur leur oxydation et gravimétrique, titrimétrique. ou gazométrique détermination des composés simples formés. éléments individuels. Premières méthodes élémentaires analyse microchimique(microanalyse) a été développée au début par F. Pregl. 20ième siècle Dès la 2ème mi-temps. 20ième siècle Pour l'analyse élémentaire des substances, l'automatique est largement utilisé. analyseurs basés sur la combustion de l'échantillon analysé org. in-va et chromato-graphique en phase gazeuse. séparation et détermination des produits de combustion. L'analyseur est équipé d'un ordinateur et automatique système d'introduction d'échantillons.
Analyse isotopique de l'org. le but est de déterminer la teneur en isotopes individuels, ainsi que de déterminer le rapport de la même organisation. des composés contenant différents ou des combinaisons de ceux-ci. À cette fin, la spectrométrie de masse ou la chromatographie multiple gaz-liquide sont le plus souvent utilisées (par exemple, lors de la séparation des formes ordinaires et deutérées du méthane ou du benzène). Naïb. la chromatographie-spectrométrie de masse est efficace.
La plupart des méthodes d'analyse fonctionnelle sont basées sur l'interaction. fonctions individuelles organisation de groupe. Connecticut. avec des réactifs adaptés. Ces districts peuvent être sélectifs ou peu sélectifs, c'est-à-dire qu'ils sont respectivement caractéristiques. seulement pour un ou plusieurs. fonctionnel groupes.
Le plus souvent, ils utilisent des solutions associées à la formation ou à la disparition de substances, de bases, d'agents oxydants, d'agents réducteurs, d'eau, de gaz et, plus rarement, de sédiments et de substances colorées. Les composés et bases résultants déterminent titrage acido-basique en milieu aquatique ou non aqueux. Dans un environnement non aqueux, des potentiomètres séparés titrage de composés et de bases de différentes concentrations lorsqu'ils sont présents ensemble.
Dans le cas de l'oxydo-réduction. solutions dont la vitesse est faible, un titrage en retour est généralement utilisé, c'est-à-dire que l'excès de réactif est titré. Sur la formation ou l'absorption de l'eau dans les quartiers de l'org. Connecticut. basé sur la définition du pluriel. fonctionnel groupes utilisant Réactif de Fischer(voir également Aquamétrie).
Les méthodes basées sur les débits, qui s'accompagnent d'un dégagement ou d'une absorption de gaz, sont rarement utilisées, car la mesure de volume ou de pression nécessite généralement un équipement volumineux.
Les mesures gravimétriques sont basées sur la formation de sédiments. méthodes pour déterminer un petit nombre de fonctions. groupes. Les composés légèrement solubles utilisés dans ces cas sont généralement métalliques. acides carbonique et sulfonique, sels org. bases, connexions complexes. (y compris ceux chélatés).
Formation de composés colorés. souvent assez spécifique et permet de déterminer sélectivement la fonction. groupes photométriques méthodes. Les solutions conduisant à la formation de composés fluorescents se sont généralisées (notamment en microanalyse), depuis la sensibilité de la détermination de la fonction. Le groupe dans ce cas est assez grand.
Un type particulier de fonctionnel. L'analyse considère des méthodes basées sur des préliminaires. interaction de la substance à déterminer avec des réactifs et détermination du produit résultant. Par exemple, aromatique après nitration, il peut être déterminé par polarographie et le rapport entre le groupe amino et le chlorure de naphtalène sulfonyle peut être déterminé par fluorimétrie.
Vous trouverez ci-dessous des exemples parmi les plus répandus. méthodes fonctionnelles fréquemment utilisées. analyse.
La détermination de l'hydrogène actif dans les alcools, les amines, les amides, les composés carboniques et sulfoniques, les mercaptans et les sulfamides repose sur leurs interactions. avec des réactifs de Grignard (généralement iodure de méthyle magnésium ; voir Méthode cérévitine)soit avec LiAlH 4 et en mesurant respectivement le volume de méthane ou d'hydrogène libéré. L'actif dans l'acétylène et ses homologues est déterminé par la solution avec les sels Ag(I), Hg(I) ou Cu(I), le dernier étant titrimétrique. détermination des séparés.
Connexions avec des insaturés les liaisons carbone-carbone sont le plus souvent bromées, iodées ou hydrogénées. Dans les deux premiers cas, Br 2 ou I 2 n'ayant pas réagi est déterminé par iodométrie et pendant l'hydrogénation, le volume de H 2 absorbé est mesuré. Le nombre de doubles liaisons peut être déterminé en ajoutant des sels de mercure à ces dernières. titrage de la substance libérée.
Lors du dosage des groupes hydroxyles, on utilise le plus souvent de l'anhydride acétique, phtalique ou pyromellitique dont l'excès est titré. Vous pouvez utiliser des chlorures d'acide. Les groupes hydroxy dans les phénols sont généralement titrés avec des solutions basiques dans un milieu non aqueux. Les phénols sont facilement bromés et combinés avec des sels de diazonium, ils sont donc titrés avec des solutions de Br 2 ou de sels de diazonium, ou un mélange bromure-bromate est ajouté à la solution étudiée, l'excès est déterminé par iodométrie (voir aussi réaction de Falin).
Les glucides peuvent être déterminés par oxydation avec du périodate de sodium et ultérieurement. titrage de l'excès d'agent oxydant ou des composés formés. De nombreux ont été développés. variantes de cette méthode (voir, par exemple, réaction de Malaprada).
Pour déterminer l'org. les composés peroxy (y compris les acides peroxy) utilisent le plus souvent leur interaction. avec KI et suivants titrage de l'I 2 libéré avec une solution de Na 2 S 2 O 3.
L'analyse des composés alcoxy consiste en une interaction. de la substance analysée avec de l'acide iodhydrique pour former des iodures d'alkyle (voir. méthode Zeisel). Ces derniers sont déterminés par différentes méthodes - gravimétriquement (sous forme d'AgI) ou titrimétriquement (titrage acide-base). Les composés carbonés peuvent être déterminés de la même manière. Pour identifier les groupes alcoxy en C 1 à C 4, les iodures d'alkyle résultants sont convertis en composés d'ammonium quaternaire, qui sont analysés par chromatographie sur couche mince ou sur papier.
La définition des groupes époxy est basée sur leur réaction avec le chlorure d'hydrogène pour former des chlorhydrines ; une fois la solution terminée, l'excès de HCl est titré avec une solution alcaline.
Pour la détermination des composés carbonylés. (aldéhydes et cétones) max. On utilise souvent l'oximation, c'est-à-dire leur conversion en interaction. avec du chlorhydrate d'hydroxylamine; Le HCl libéré à la suite de la réaction est titré avec une solution alcaline (le point final du titrage est fixé à l'aide d'un indicateur ou de manière potentiométrique). Il existe un grand nombre de modifications de cette méthode. Les aldéhydes peuvent également être déterminés par la solution avec du bisulfite de Na suivie de. titrage acido-basique. Les aldéhydes avec les ions Ag +, la réaction avec les hydrazines et la formation de bases de Schiff sont moins couramment utilisés.
Les quinones sont réduites avec du chlorure de Ti(III) ou du sulfate de V(II) ; l'excès d'agent réducteur est déterminé par titrimétrie. Les quinones peuvent également être déterminées par iodométrie.
Pour déterminer les composés carbonés et leurs sels, max. Le titrage acide-base est souvent utilisé dans des milieux non aqueux.
Un grand nombre de méthodes ont été développées pour l'analyse des dérivés carbonés. Les anhydrides après leur hydrolyse en solution sont titrés avec des solutions alcalines. Dans le cas de l'analyse d'un mélange d'un acide et de son anhydride, la somme des deux substances est déterminée par titrage acido-basique, puis l'anhydride est mélangé avec de la morpholine ou de l'aniline et les acides libérés sont titrés. Dans ce dernier cas, vous pouvez également déterminer l'excès de base par titrage avec une solution de HCl. Les halogénures d'acide ou leurs mélanges avec des composés sont déterminés de la même manière. Dans ce cas, au lieu de solutions avec des amines, des interactions sont souvent utilisées. halogénure d'acide avec de l'alcool et enfin. titrage séparé du libre l'acide carbonique et l'acide riche en halogène libéré avec une solution alcaline.
Le dosage des esters carbonés repose sur leur hydrolyse avec une solution alcaline, l'excès est titré avec une solution. De petites quantités d'esters sont généralement déterminées par spectrophotométrie sous forme de sels de Fe(III) d'acides hydroxamiques formés lors de l'interaction. esters avec l'hydroxylamine.
Pour déterminer l'org. un grand nombre de méthodes ont été proposées. Les composés capables de réduction (nitro-, nitroso-, ) sont déterminés par voie titane ou vanadatométrique : une solution en excès de sel de Ti(III) ou de sel de V(II) est ajoutée et l'agent réducteur n'ayant pas réagi est titré avec une solution de Fe(III) sel.
Le titrage d'une solution de ramie (généralement HClO 4) dans un milieu non aqueux est largement utilisé dans la détermination. Cette méthode vous permet souvent de déterminer séparément l'organisation. et non-org. bases en mélanges, ainsi que org. bases de force variable lorsqu’elles sont présentes ensemble. Les amines peuvent être déterminées, comme les dérivés hydroxylés, par le rapport de leur acylation. Déterminer les aromatiques primaires. les amines sont souvent titrées en solution en milieu acide, accompagnées de la formation d'un composé diazoïque. Un titrage similaire des amines secondaires conduit à leur N-nitrosation et est également utilisé dans l'analyse. Lors de la microanalyse des aromatiques primaires. amines, les composés diazoïques résultants sont généralement combinés avec les composants azoïques correspondants et le colorant résultant est déterminé par spectrophotométrie. Dans le cas de l'analyse de mélanges d'amines primaires, secondaires et tertiaires, le titrage avec une solution de HClO 4 est le plus souvent utilisé en milieu non aqueux du mélange initial (toutes les amines sont titrées), le mélange après acétylation avec de l'anhydride acétique (uniquement tertiaire les amines sont titrées) et le mélange après traitement à l'acétylacétone ou à l'aldéhyde salicylique (la somme des amines secondaires et tertiaires est titrée).
Pour déterminer les sels d'aryldiazonium avec une solution de la substance analysée, titrez les portions pesées du composant azoïque (3-méthyl-1-phényl-5-pyrazolone, m-phénylènediamine, etc.) ou ajoutez une solution du composant azoïque à l'analyse. solution, l'excédent à L'essaim est titré avec une solution de NaNO 2 en milieu acide. Dans le cas de l'analyse des composés diazoïques, il est également possible d'utiliser la gazométrie. analyse basée sur la décomposition du composé étudié. avec libération de N 2 dont le volume est mesuré. Parfois, comme dans le cas de l'analyse des amines, les composés diazoïques sont déterminés par la combinaison avec ces derniers. riche en spectrophotométrie. détermination du colorant obtenu.
Les hydrazines sont généralement titrées par voie iodométrique. Dans le cas des thiols, l'interaction peut également être utilisée. avec des sels d'argent ou un titrage acido-basique. Org. les sulfures sont oxydés avec un mélange bromure-bromate, l'excès est dosé par titrimétrie.
Répandu pour la qualité. et les quantités. fonctionnel Des méthodes sélectives et assez sensibles de spectroscopie IR et RMN ont également été analysées.
L’émergence d’une analyse stéréospécifique de l’org. en-en 2ème mi-temps. 20ième siècle associé au développement de la chromatographie méthodes. Pour séparer les énantiomères, on effectue le plus souvent une réaction préalable entre les substances analysées et des réactifs optiquement actifs pour former des diastéréomères, qui sont ensuite séparés par chromatographie gaz-liquide ou liquide haute performance sur des colonnes à phases stationnaires optiquement actives.
Analyse moléculaire de l'org. in-in fondé ch. arr. sur l'utilisation de la chromatographie et autres. les méthodes spectrales, qui permettent d'établir la structure de l'org. Connexions.
Analyse de phase, qui permet une analyse qualitative et quantitative des matériaux cristallins. formulaires d'organisation connexion, réalisée à l'aide radiographie Et électronique. Radiographie, analyse structurelle vous permet de définir la structure structurelle de l’organisation avec une grande précision. c., déterminer les longueurs des liaisons entre les atomes et les angles entre les liaisons.
Les méthodes d'analyse énumérées ci-dessus sont basées sur la détermination directe des substances analysées ou des dérivés obtenus à partir de celles-ci. Dans O. c. UN. Les méthodes indirectes sont également souvent utilisées. Ainsi, par exemple, les composés carbonés peuvent être isolés du mélange analysé sous forme d'argent ou d'autres sels peu solubles, puis en utilisant la méthode d'absorption atomique. spectroscopie ou analyse par fluorescence X pour déterminer la quantité du métal correspondant ; Sur la base des résultats d'une telle analyse, la teneur en dioxyde de carbone peut être calculée. En chromatographie liquide, l'utilisation de la détection indirecte des substances séparées est efficace, dans laquelle un composant actif est ajouté à la phase mobile, formant des composés facilement détectables avec les produits de séparation ou avec les substances chromatographiées.
Les méthodes d'analyse et le matériel utilisé dépendent de la tâche spécifique d'O. v. a. : détermination de l'ingrédient principal du mélange, org. ou non-organisation. impuretés dans l'org. wow, org. impuretés inorganiques in-ve ou analyse d'un mélange complexe à plusieurs composants d'in-in.
Méthodes O. siècle. UN. largement utilisé dans le développement de la technologie industrielle. produit par org. produits et dans le processus de production lui-même pour le développement de méthodes d'analyse des matières premières, auxiliaires. dedans, entre les deux. produits à différentes étapes de production, pour contrôler la production. processus, produits finis, eaux usées et émissions de gaz, pour identifier les impuretés dans les produits intermédiaires et finaux et pour développer des analytes. techniques qui assurent la cinétique nécessaire. recherche. Dans tous les cas, il faut choisir celui qui est optimal. options pour les méthodes d'analyse et leurs combinaisons conformément aux exigences de rapidité, de reproductibilité, de précision, etc.
Lors du développement d’un analyte. parties normatives et techniques. documentation pour les matières premières, auxiliaire. les matériaux et les produits finis établissent tout d'abord le nombre minimum nécessaire et suffisant d'analytes. indicateurs. Ces indicateurs incluent le point de fusion, le pH et la teneur en base. substances contenues dans le produit, qui sont déterminées par une méthode directe (généralement par titrimétrie utilisant la potentiométrie) ou indirectement, en soustrayant de la masse du produit entier la masse d'impuretés déterminée par chromatographie. (le plus souvent), électrochimique. ou spectrophotométrique. méthodes. Lors de l'utilisation de la fonction. analyse pour déterminer les principaux les éléments choisissent généralement une méthode qui consiste à déterminer cet élément par fonction. groupe constitué à la dernière étape de sa réception. Si nécessaire, lorsque la substance analysée est obtenue par synthèse en plusieurs étapes, elle est déterminée selon différentes fonctions. groupes. Analyste. les méthodes choisies pour l'analyse des matières premières et des produits finis doivent avoir le Ch. arr. bonne reproductibilité et précision.
Les méthodes analytiques utilisées dans le contrôle de la production doivent être rapides et continues (par exemple, redox-métrie, pH-métrie, ). Les méthodes de suivi des processus de production sont basées sur l'org. in-in est souvent la définition d'une fonction qui disparaît. groupe, c'est-à-dire un groupe en transformation à une étape donnée de la production, ce qui permet d'enregistrer avec précision la fin de l'étape correspondante. Dans ce cas, la chromatographie liquide à haute performance, la spectrophotométrie et l'électrochimie en couche mince sont largement utilisées. méthodes, flux-injection. analyse.
Pour l'analyse, il y aura des intervalles. la titrimétrie est le plus souvent utilisée pour les produits manufacturés et pour l'analyse des réactions. mélanges-chromatographiques complexes. et des méthodes spectrales, y compris la chromatographie en phase gazeuse-spectrométrie de masse, une combinaison de chromatographie en phase gazeuse et de spectroscopie infrarouge à transformée de Fourier.
L'analyse des objets environnementaux a acquis une grande importance. Lors du développement de méthodes appropriées d'analyse de base. leurs exigences sont une sensibilité élevée et une identification correcte des substances déterminées. Ces exigences sont satisfaites par la chromatographie en phase gazeuse-spectrométrie de masse utilisant deux ou plusieurs phases stationnaires.
En clinique analyse (analyse du sang, de l'urine, des tissus et d'autres objets pour le contenu de médicaments, métabolites, stéroïdes, acides aminés, etc.) est importante non seulement la sensibilité, l'exactitude et la rapidité de l'analyse, mais également la reproductibilité de ses résultats. Lorsque cette dernière exigence est critique, la chromatographie en phase gazeuse-spectrométrie de masse dans des conditions standard, ainsi que l'injection en flux à haut débit, sont utilisées. analyse et une variété de méthodes enzymatiques avec une sélectivité élevée.
Lit. : Guben Weil, Méthodes de chimie organique, vol 2, Méthodes d'analyse, trans. avec lui. 4e éd., M.. 1963 ; Cheronis N. D., Ma T. S., Méthodes micro- et semi-micro d'analyse fonctionnelle organique, trans. de l'anglais, M., 1973 ; Siggia S. Hannah J. G., Analyse organique quantitative par groupes fonctionnels, trans. de l'anglais, M. ; 1983. B. JE. Kolokolov.
Encyclopédie chimique. - M. : Encyclopédie soviétique. Éd. I.L. Knunyants. 1988 .
Voyez ce qu'est « ANALYSE DE SUBSTANCES ORGANIQUES » dans d'autres dictionnaires :
L'analyse de l'eau est une méthode permettant d'étudier les propriétés et les qualités de l'eau. Il est utilisé pour déterminer la quantité de diverses substances dans l’eau qui est en contact avec les humains à des fins industrielles et domestiques ou à des fins scientifiques. Table des matières 1 Types d'eau pour ... ... Wikipedia
L'analyse du sol est un ensemble d'opérations réalisées pour déterminer la composition, les propriétés physico-mécaniques, physico-chimiques, chimiques, agrochimiques et biologiques du sol. Réaliser des travaux mécaniques (granulométriques), chimiques, ... ... Wikipedia
ANALYSE DE L'EAU- est réalisée pour déterminer la qualité de l'eau et déterminer la possibilité de l'utiliser pour alimenter les étangs piscicoles. Un V. a lieu quatre fois par an : au printemps (pendant la crue printanière), au milieu de l'été (juillet), en automne (pendant l'automne... ... Pisciculture en étang
analyse- L'ANALYSE (du grec analyse, décomposition, démembrement) est la procédure de division réelle ou mentale d'un objet, d'un phénomène ou d'un processus, ainsi que de leurs relations en composants, éléments, propriétés, fonctions et sous-systèmes. La procédure... ... Encyclopédie d'épistémologie et de philosophie des sciences
Identification (détection) des composants analysés en quantités approximatives, évaluation de leur teneur dans les eaux et les matériaux. En tant que composants, il peut s'agir atomes et ions, isotopes d'éléments et nucléides individuels, molécules, fonctionnels. groupes et radicaux... Encyclopédie chimique
Détermination du contenu (masse, concentration, etc.) ou des quantités. ratios de composants dans l’échantillon analysé. Des composants déterminés peuvent l'être. atomes, molécules, isotopes, fonctionnels. groupes, phases, etc. (voir Analyse élémentaire, Moléculaire... ... Encyclopédie chimique
Établissement d'enseignement « Université d'État de Brest du nom d'A.S. Pouchkine"
Département de Chimie
TRAVAIL DE COURS
Méthodes d'étude des composés organiques
Effectué :
Étudiant de 5ème année,
Faculté de biologie
spécialité « Biologie. Chimie"
éducation à plein temps
Petruchik Irina Alexandrovna
Conseiller scientifique:
Borichevsky
Alexandre Ivanovitch
Brest, 2012
Méthodes d'étude des composés organiques
TABLE DES MATIÈRES
INTRODUCTION……………………………………………………………….. 3
- Classification des méthodes d'étude des substances organiques………. 4
Les méthodes les plus simples pour étudier les substances organiques
2.1.1 Cristallisation………………………………………… ……… 6
2.1.2 Sublimation…………………………………………………………. 7
2.1.3 Distillation……………………………………………………….. 8
2.1.4 Chromatographie…………………………………………… …. 9-11
2.2 Analyse des substances organiques………………………………….. 12-13
- Méthodes physicochimiques pour l'étude des substances organiques... 14
3.2 Calorimétrie……………………………………………………………… ……… 17
3.3 Diffraction des rayons X et électronique…………………………… 18-19
3.4 Méthodes de recherche électrochimique…………………… 20-21
3.5 Spectroscopie…………………………………………… …….. 22-27
CONCLUSION…………………………………………………… ……….…. 28
LISTE DE RÉFÉRENCES…………………………. 29
INTRODUCTION
L'étude des substances organiques vise à établir la structure de la substance, sa structure spatiale et ses orbitales moléculaires caractéristiques, à étudier l'interaction des atomes et des molécules et à étudier les vitesses et les mécanismes des réactions. En raison du grand nombre de composés organiques différents, il est impossible de développer un schéma analytique unique, comme cela se fait souvent dans l'analyse quantitative inorganique. Et pourtant, des recherches systématiques permettent d'identifier la matière organique de manière assez fiable et rapide.
L'établissement de la structure de la matière organique est l'objectif principal de leur étude, quelle que soit la méthode de recherche. Cependant, les intérêts liés à l'étude de l'un ou l'autre composé organique sont déjà d'une autre nature. Les questions liées aux ressources naturelles de notre planète revêtent une importance particulière. Nous savons que les sources de pétrole et de gaz revêtent une importance particulière pour l’humanité, mais elles sont limitées. Par conséquent, le problème de la recherche de nouvelles matières premières pour la synthèse organique et pétrochimique et de la production artificielle de pétrole et de gaz est devenu urgent. Mais ce n’est là qu’une des raisons qui justifient l’étude de la matière organique. Si vous regardez autour de vous, toute vie sur Terre est issue de la chimie organique. En conséquence, l'étude des substances organiques est la clé des découvertes mondiales dans le domaine de la nature vivante, la possibilité d'apprendre tous les processus vitaux, de trouver des moyens de guérir de nombreuses maladies terribles, de créer nous-mêmes de la matière vivante, etc.
- Classification des méthodes d'étude des substances organiques.
- les méthodes d'étude les plus simples : purification des substances organiques (cristallisation, sublimation, distillation, chromatographie, filtration sur gel, électrophorèse) et analyse des substances organiques (analyses élémentaires quantitatives et qualitatives) ;
- méthodes physiques et chimiques : réfractométrie, calorimétrie, mesure des moments dipolaires électriques, radiographie et diffraction électronique, méthodes électrochimiques (polarographie, voltammétrie anodique), spectroscopie (photoélectron, spectroscopie de masse, infrarouge, etc.)
Les méthodes les plus simples pour étudier les substances organiques
- Épuration biologique
Pour caractériser la pureté d'une substance, les constantes et méthodes suivantes sont utilisées : point de fusion, température de cristallisation, point d'ébullition, indice de réfraction de la lumière, densité, données de spectres d'absorption (coefficient d'intensité d'absorption dans les spectres électronique et infrarouge), résonance magnétique nucléaire (RMN). ) données spectrales, spectrométrie de masse, analyse chromatographique, analyse luminescente, etc.
Obtenir une substance pure signifie séparer un mélange donné de substances en substances individuelles et les purifier jusqu'au degré de pureté souhaité. Il faut ici distinguer deux ensembles de méthodes : les méthodes de séparation d'un mélange en composants qui ne sont pas encore purs et les méthodes de purification finale.
Lorsqu’on parle de la pureté des substances chimiques, il faut savoir qu’une substance absolument pure ne peut être imaginée que théoriquement. Il n’existe pas et il ne peut y avoir de substances absolument pures. Selon la méthode de purification, la substance contient une certaine quantité d'impuretés. Les méthodes de purification conventionnelles peuvent atteindre une teneur en substance principale de 99,9 à 99,95 %. Grâce à des méthodes spéciales de nettoyage en profondeur, il est possible de réduire la teneur en impuretés des substances organiques à 10 -3 ....10 -4 %
2.1.1 Cristallisation
La cristallisation est une méthode classique de purification des substances cristallines. La méthode est basée sur le fait que différentes substances ont des solubilités différentes dans un certain solvant et qu'une diminution de la température (à de rares exceptions près) entraîne une diminution de la solubilité des substances. En filtrant la solution chaude, les impuretés insolubles sont séparées et après refroidissement, la substance est libérée de la solution sous forme de cristaux. Des recristallisations répétées réduisent généralement la quantité d'impuretés. Une variante du procédé est la cristallisation en fusion. Une option spéciale – la fusion de zone – est utilisée pour une purification en profondeur des substances.
Par exemple : nous devons nettoyer l'acide salicylique des impuretés. Pour ce faire, nous prenons une masse pré-pesée de cet acide et calculons le volume requis de solvant - eau, afin d'obtenir une solution saturée, qui peut ensuite être cristallisée.
2.1.2 Sublimation (Sublimation)
De nombreuses substances cristallines ont la capacité de se sublimer, c'est-à-dire au passage à la phase gazeuse, en contournant la phase liquide, suivi d'une cristallisation à partir de la phase gazeuse. Cette méthode permet de séparer les substances sublimantes des impuretés non sublimantes et de séparer un mélange de substances avec différentes températures de sublimation ou températures de cristallisation de la phase gazeuse (sublimation dégradée). Si les substances sont difficiles à sublimer et à se décomposer à haute température, la sublimation est utilisée sous vide ou sous vide poussé - jusqu'à 0,0013 Pa (10 -5 mm Hg ; 1 mm Hg = 133,3 Pa). La sublimation sous vide poussé dans différentes versions est utilisée pour un nettoyage en profondeur.
La purification d'un solide par sublimation n'est possible que si sa pression de vapeur est supérieure à la pression de vapeur des impuretés. Lorsque la pression de vapeur du solide correspond à la pression appliquée, les meilleurs résultats sont obtenus.
Par exemple : l'E-stilbène est sublimé à une température de 100 o C et une pression de 20 mm Hg. Art.
2.1.3 Distillation (distillation)
Pour de nombreuses substances à bas point de fusion et la plupart des liquides, une bonne méthode de nettoyage consiste à
Distillation fractionnée, à condition que la différence de points d'ébullition des composants du mélange soit suffisamment grande et qu'aucun mélange azéotropique ne se forme. La sélectivité (efficacité) de la distillation fractionnée peut être augmentée à l'aide de dispositifs spéciaux : condenseurs à reflux, colonnes de distillation, etc. Pour les substances à point d'ébullition élevé, la distillation sous vide est utilisée. Une variante de la méthode est la distillation de systèmes à deux composants, qui se séparent lorsqu'ils sont refroidis, par exemple la distillation à la vapeur : le limonène (point d'ébullition 178 o C à 760 mm Hg) est distillé avec de l'eau (point d'ébullition 100 o C à 760 mm Hg. Art.) à une température de 98°C. Dans ce cas, le rapport quantitatif dans le distillat (en grammes) de limonène : eau est de 1 : 1,54.
2.1.4 Chromatographie
Les méthodes de séparation chromatographique sont basées sur la capacité différente des substances à être adsorbées à la surface du sorbant ou réparties entre deux phases non miscibles (liquide-liquide, liquide-gaz), dont une phase (liquide) est située à la surface du absorbant. Il existe donc différents types de chromatographie, à savoir : la chromatographie d'adsorption et de partage liquide, la chromatographie en phase gazeuse.
La chromatographie d'adsorption liquide est basée sur la capacité différente des substances à être sorbées à la surface du sorbant et désorbées lors du passage d'un solvant-éluant. Comme sorbants, on utilise de l'oxyde d'aluminium, de l'acide silicique et du dioxyde de silicium (gels de silice), des polysaccharides granulaires (dextranes) ou d'autres polymères qui gonflent dans un solvant, formant un gel granulaire (chromatographie sur gel).
La chromatographie de partage liquide est un type de chromatographie d'adsorption dans laquelle le sorbant (support) est recouvert d'un mince film de liquide. L'éluant est généralement un solvant qui ne se mélange pas au liquide présent sur le sorbant. Lors du passage de l'éluant, des substances sont réparties entre la phase liquide et l'éluant. Ce type de chromatographie est particulièrement adapté à la séparation de substances hautement solubles dans l'eau ou capables de former des sels solubles dans l'eau. Ces substances comprennent le sucre, les acides aminés, de nombreux colorants organiques, la plupart des alcaloïdes, les acides mono- et polycarboxyliques, les alcools, etc.
Un exemple de chromatographie liquide d'un mélange d'étalons de phospholipides synthétiques (1) et d'un échantillon d'un extrait lipidique brut de la membrane cellulaire d'érythrocytes humains (2) sur une colonne de phase normale lorsqu'il est détecté avec un détecteur de diffusion de lumière laser.NL - neutre lipides; PE – phosphatidyléthanolamine ; PS – phosphatidylsérine ; PC – phosphatidylcholine ; SM – sphingomyéline.
La chromatographie en phase gazeuse est utilisée pour séparer des mélanges de substances liquides et solides gazeuses ou volatiles. Le principe de la méthode est similaire à celui de la chromatographie liquide. Le mélange à séparer est dilué avec un gaz vecteur (H 2, N 2, He) et introduit dans des colonnes d'adsorption. Le gaz vecteur est à la fois un solvant et un éluant. De fines poudres de matériaux silicatés, qui peuvent être pures (chromatographie par adsorption gazeuse) ou recouvertes d'un film de liquide non volatil (chromatographie gaz-liquide), sont utilisées comme sorbants. Des capillaires recouverts à l’intérieur d’un film de liquide non volatil (chromatographie capillaire) sont également utilisés. Le gaz vecteur désorbe progressivement les composants du mélange et les emporte avec lui. La présence de substances organiques dans le gaz vecteur et leur quantité sont détectées à l'aide de détecteurs spéciaux et enregistrées par un enregistreur. En chromatographie préparative, le gaz porteur passe ensuite à travers des récepteurs spéciaux dans lesquels les substances organiques sont capturées par congélation.
Cette méthode permet d'obtenir une séparation complète du mélange. Lors de l'utilisation de colonnes d'adsorption de haute puissance, la méthode est utilisée comme méthode préparative pour la séparation de petites quantités de substances (1...10 g).
Exemple de chromatographie en phase gazeuse : analyse à grande vitesse de vapeurs explosives sur colonne polycapillaire à une température de 170°C.
Une colonne polycapillaire d'une longueur de seulement 22 cm permet de détecter et d'identifier des traces de vapeurs explosives en 2,5 minutes : 1 - 2,6-dinitrotoluène, 2 - 2,4-dinitrotoluène. 3 - 2,4,6-trinitrotoluène, 4 - 3,4,5-trinitrotoluène, 5 - 2,3,4-trinitrotoluène, 6 - hexogène. 7 - tétryl.
- Analyse de la matière organique
La première tâche est la détermination qualitative et quantitative de la composition élémentaire. Ensuite, selon les données d'analyse élémentaire, la formule récapitulative la plus simple est calculée, le poids moléculaire est déterminé et la véritable formule moléculaire brute est calculée. Enfin, la dernière étape consiste à déterminer la structure moléculaire. A cet effet, des méthodes chimiques sont utilisées (clivage progressif, dérivation), et récemment des méthodes physico-chimiques (spectroscopie de masse, analyse par diffraction des rayons X, spectroscopie) sont de plus en plus utilisées.
Analyse élémentaire quantitative et qualitative
Les méthodes analytiques sont basées sur la dégradation complète de la matière organique résultant d'une oxydation ou d'autres moyens et sur la détermination d'éléments chimiques à l'aide de méthodes connues. Le carbone est dosé sous forme de CO 2, l'hydrogène - sous forme de H 2 O, l'azote - en mesurant le volume de N 2 ou en dosant NH 3 ou NaCN (selon le type de décomposition), les halogènes - sous forme de ions halogénures, soufre - sous forme d'ions sulfate ou sulfure, phosphore signifiant ion phosphate, etc.
Le carbone et l'hydrogène sont dosés qualitativement par chauffage avec CuO :
CnH2n+3nCuO>nCO2+nH2O+3nCu
Et le monoxyde de carbone libéré est détecté en faisant passer le gaz dans la solution de Ba(OH) 2, et l'eau est détectée visuellement sur les parois du tube à essai.
L'azote, le soufre et les halogènes sont déterminés qualitativement par fusion avec le sodium. Les halogénures de NaCN, Na 2 S et de sodium résultants sont détectés en solution aqueuse par des réactions analytiques conventionnelles.
Il existe des échantillons spéciaux pour l'analyse quantitative des composés organiques. Auparavant, des installations de macroanalyse étaient généralement utilisées (l'échantillon pesait 0,2 ... 0,5 g). De nos jours, divers appareils sont courants pour la microanalyse (échantillon 0,001 ... 0,01 g), pour l'ultramicroanalyse (échantillon 10 -5 ... 10 -4 g). Pour la détermination quantitative du carbone et de l'hydrogène, on utilise des appareils dans lesquels la matière organique est brûlée dans un flux d'oxygène : le CO 2 est capté avec une solution de KOH, et le H 2 O avec un absorbant spécial et déterminé par pesée. Pour quantifier l'azote, la substance est brûlée lorsqu'elle est chauffée avec CuO et le volume de gaz libéré est mesuré dans un azomètre sur une solution de KOH. Les halogènes et le soufre sont quantifiés en brûlant l'échantillon dans une atmosphère d'oxygène, en dissolvant les gaz dans l'eau et en titrant les ions halogénures ou les ions sulfate.
Des microanalyseurs automatiques ont été développés en utilisant le principe de la chromatographie en phase gazeuse, dans laquelle le carbone, l'hydrogène, l'azote et le soufre sont déterminés simultanément.
Le poids moléculaire d'un composé est généralement déterminé par spectrométrie de masse.
- Méthodes physicochimiques pour l'étude des substances organiques
- Méthodes spectrales et autres méthodes optiques ;
Méthodes électrochimiques ;
Méthodes d'analyse chromatographiques.
Un groupe de méthodes d'analyse électrochimiques, basées sur la mesure de la conductivité électrique, des potentiels et d'autres propriétés, comprend les méthodes de conductométrie, de potentiométrie, de voltamétrie, etc.
Mais afin de vérifier avec précision la meilleure efficacité de ces méthodes et leur grande importance pratique réelle, considérons à des fins de comparaison d'autres méthodes physico-chimiques.
- Réfractométrie
, où n est l'indice de réfraction de la lumière pour la raie D du sodium (589 nm) ; M est le poids moléculaire de la substance ; ?? - densité.
La réfraction moléculaire a des propriétés additives, c'est-à-dire la réfraction moléculaire d'une molécule peut être obtenue en additionnant les réfractions des éléments constitutifs de la molécule. Ces composants sont des liaisons chimiques et un ensemble de liaisons et d'atomes. Ces réfractions sont calculées à partir d'études sur de nombreux composés organiques et peuvent être trouvées dans des ouvrages de référence. Par exemple:
R CH4 = 4 R C-H ; R CH3NO2 = 3 R C-H + R C-N + R NO2
Le phénomène de réfraction de la lumière est associé à la polarisabilité du système électronique de molécules. Sous l'influence du champ électromagnétique de la lumière, la polarisation des molécules se produit, principalement de leurs systèmes électroniques. Plus le système électronique d'une molécule est mobile, plus l'indice de réfraction de la lumière et la réfraction moléculaire sont élevés.
Les études de réfraction moléculaire peuvent être utilisées pour déterminer la structure d'un composé. Ainsi, pour le composé étudié, la réfraction moléculaire est déterminée expérimentalement et comparée à la réfraction obtenue en sommant les réfractions des liaisons selon la formule développée proposée. Si les résultats coïncident, alors la structure peut être considérée comme prouvée ; sinon, nous devons alors chercher une autre structure ; Dans certains cas, une forte augmentation de la réfraction moléculaire est observée par rapport à celle attendue (exaltation de la réfraction). Ceci est typique des systèmes couplés.
Les valeurs de réfraction moléculaire des liaisons chimiques, des atomes, des molécules et des ions peuvent être utilisées pour évaluer qualitativement leur polarisabilité. La polarisabilité d'une molécule (ion, liaison) est sa capacité à se polariser, c'est-à-dire à un changement de la position des noyaux et de l'état du nuage électronique sous l'influence d'un champ électrique externe. Il se produit principalement une polarisation électronique.
3.2 Calorimétrie
La calorimétrie est une méthode permettant d'étudier les effets thermiques des réactions chimiques et des processus de transition de phase (par exemple, fusion, cristallisation, sublimation, condensation). Le processus (réaction) est effectué dans des appareils spéciaux - des calorimètres et la chaleur dégagée ou absorbée est quantifiée.
Les chaleurs molaires de combustion des substances sont déterminées par calorimétrie. À son tour, la chaleur de combustion (W) est utilisée pour calculer la chaleur de formation d'une substance E ou l'enthalpie standard de formation H 0 ? La chaleur de formation d'une substance peut être calculée à partir d'éléments à l'état atomique ou d'éléments à l'état « standard » (carbone sous forme de graphite, hydrogène gazeux, etc.), et les valeurs numériques qui en résultent, bien entendu, différer. Lorsque vous examinez des données tabulaires, vous devez y prêter une attention particulière. En règle générale, les chaleurs de formation de substances pour un processus sont calculées à partir des atomes des éléments et ?H 0 - à partir des éléments à l'état « standard ». Par exemple, la chaleur de formation d'hydrocarbures à partir d'atomes :
- nS - ] – W, où W est la chaleur de combustion ; - chaleur de formation du CO 2 (393,5 kJ/mol) ; - chaleur de formation de l'eau (285,8 kJ/mol) ; S – chaleur d'atomisation (sublimation) du carbone (graphite) (-715 kJ/mol) ; - chaleur d'atomisation (dissociation) d'une molécule d'hydrogène (-436 kJ/mol).
Plus la chaleur de combustion est faible, plus la chaleur de formation de composés de même composition est grande.
Cette méthode sert principalement à comparer et caractériser la stabilité et la réactivité des composés organiques.
3.3 Radiographie et diffraction électronique
La méthode des rayons X - analyse par diffraction des rayons X - est basée sur la diffraction des rayons X dans un cristal d'une substance. Les rayons X (rayonnement électromagnétique d'une longueur d'onde de 0,1 à 10 nm) lorsqu'ils traversent un cristal interagissent avec les coques électroniques des atomes. À la suite de cette interaction, une diffraction des rayons X se produit et un diagramme de diffraction est obtenu sur un film photographique - des taches ou des cercles. À partir du diagramme de diffraction, à l'aide de calculs complexes, des informations sont obtenues sur le placement des molécules dans la cellule unitaire d'un cristal et sur les distances entre les atomes et les angles entre les liaisons chimiques. Plus le nombre d’électrons dans un atome est petit, plus les réflexions des rayons X sont faibles. Il est donc très difficile de déterminer l’emplacement des atomes d’hydrogène.
La méthode de diffraction électronique est similaire à la méthode des rayons X et repose sur l'interaction d'un flux d'électrons avec une substance. Le flux d’électrons traversant une substance ressemble à un rayonnement électromagnétique de très courte longueur d’onde et produit un diagramme de diffraction. Ces diagrammes de diffraction (diagrammes de diffraction électronique) peuvent être obtenus pour des substances à l'état gazeux ou pour des films très minces. La diffraction électronique est provoquée par l'interaction des électrons avec les noyaux atomiques.
Ces méthodes d'analyse structurale permettent de déterminer la structure complète de la molécule - distances interatomiques, angles entre liaisons, c'est-à-dire la disposition spatiale exacte de tous les atomes d'une molécule dans un réseau cristallin ou à l'état gazeux. La structure de substances naturelles complexes telles que le saccharose, la pénicilline, la strychnine, la vitamine B 12, certaines protéines (myoglobine) et les acides nucléiques a été déterminée par analyse par diffraction des rayons X.
À partir des méthodes de recherche aux rayons X, il a été constaté que le rayon covalent des atomes lors de l'hybridation sp 2 - et sp varie en fonction du type de liaison, par exemple dans la double liaison C=C (C sp2 - C sp2), le rayon covalent de l'atome de carbone C sp2 est inférieur à celui des liaisons =C-C (C sp2 - C sp3). Dans le 1er cas, il est de 0,067 nm, dans le 2ème - 0,076 nm et dans le cas du benzène - 0,0695 nm, c'est-à-dire La longueur des liaisons dépend également du composé lui-même, et pour chaque composé, les longueurs des liaisons constituent une caractéristique individuelle, qui peut être utile pour identifier un composé organique spécifique.
3.4 Méthodes de recherche électrochimique
Les méthodes électrochimiques sont basées sur la dépendance de l'intensité du courant sur la tension appliquée lorsque le courant traverse une solution dans des électrolyseurs spécialement conçus. En conséquence, des courbes courant-tension (potentiel) apparaissent. Ces courbes courant-tension caractérisent les processus se déroulant sur les électrodes. La réduction électrochimique se produit au niveau de la caote et l'oxydation électrochimique se produit au niveau de l'anode. Selon le type de procédé étudié (anode ou cathodique), on utilise des dispositifs qui diffèrent par le rapport entre les surfaces d'électrode, le matériau de l'électrode, etc.
Polarographie
La méthode polarographique est basée sur des processus cathodiques (l'ajout d'un électron à une substance sur une électrode à mercure). Le schéma de circuit d’un polarographe est très simple. Il se compose d'une microélectrode de mercure dégoulinante avec une surface continuellement renouvelée et d'une électrode de référence (mercure ou autre électrode normale). La surface cathodique est nettement plus petite que la surface anodique, les processus de polarisation cathodique sont donc décisifs dans ce cas. La substance organique diffuse vers la cathode et accepte un électron, et la cathode se dépolarise. La dépolarisation de la cathode commence à un certain potentiel E ext (potentiel de réduction ou de libération caractéristique d'un dépolariseur donné. En conséquence, l'électrolyse commence et l'intensité du courant augmente fortement. Avec une augmentation progressive de la tension, une certaine valeur stationnaire de l'intensité du courant (courant limite) est établi, qui ne dépend plus de l'augmentation de la tension.
La polarographie peut être utilisée pour caractériser le processus :
La méthode de polarographie est largement utilisée pour déterminer la concentration de substances dans des solutions.
Voltamétrie anodique
Cette méthode est basée sur des procédés anodiques (oxydation d'un composé organique sur une anode en platine ou en graphite). D'un point de vue expérimental, cette méthode s'apparente à la polarographie.
La voltamétrie anodique est utilisée pour étudier les processus d'oxydation :
La méthode est également utilisée pour la détermination quantitative de substances dans des solutions.
3.5 Spectroscopie
Les méthodes spectroscopiques sont basées sur l'interaction d'une substance avec un rayonnement électromagnétique, qui provoque l'absorption du rayonnement ou son émission. L'interaction est possible dans une très large gamme d'ondes électromagnétiques, depuis les rayons ? jusqu'aux ondes radio.
Selon la région du spectre électromagnétique, diverses méthodes et instruments expérimentaux sont utilisés.
Les champs de rayonnement électromagnétique les plus couramment utilisés en chimie organique sont :
- la région ultraviolette (UV) et visible du spectre, où l'énergie nécessaire pour exciter les électrons de la molécule est absorbée (un type de spectroscopie électronique) ;
- la région infrarouge (IR), où est absorbée l'énergie nécessaire au changement des états vibrationnels de la molécule (spectroscopie vibrationnelle) ;
- la région du rayonnement radiofréquence où l'énergie est dépensée pour réorienter les spins des noyaux (spectroscopie de résonance magnétique nucléaire - RMN).
Les méthodes spectrales sont utilisées pour identifier et établir la structure des composés, analyser des mélanges et permettre également de suivre l'évolution des transformations chimiques. L'avantage des méthodes spectrales est la faible consommation de substance (1 mg ou moins).
Spectroscopie électronique
Le spectre électronique apparaît lorsqu'une substance absorbe le rayonnement ultraviolet (longueurs d'onde 22 à 400 nm) et visible (400 à 800 nm). Il n'y a pas de différence fondamentale entre ces parties du spectre ; elles diffèrent uniquement par le fait que des ondes d'une longueur de 400 à 800 nm sont perçues par l'œil humain et que nous voyons la substance colorée.
Sous l'influence de la lumière UV, la molécule est excitée, c'est-à-dire transition des électrons vers un niveau plus excité et redistribution de la densité électronique dans la molécule. Les électrons formant les liaisons ? sont les plus difficiles à exciter ; les électrons des liaisons ? et les paires isolées d'électrons sont plus faciles à exciter.
Envoyer votre bon travail dans la base de connaissances est simple. Utilisez le formulaire ci-dessous
Les étudiants, étudiants diplômés, jeunes scientifiques qui utilisent la base de connaissances dans leurs études et leur travail vous en seront très reconnaissants.
Posté sur http://www.allbest.ru/
Caractéristiques de l'analyse des composés organiques :
Les réactions avec les substances organiques se déroulent lentement avec formation de produits intermédiaires.
Les substances organiques sont thermolabiles et se carbonisent lorsqu'elles sont chauffées.
L'analyse pharmaceutique des substances médicinales organiques repose sur les principes de l'analyse fonctionnelle et élémentaire.
Analyse fonctionnelle - analyse par groupes fonctionnels, c'est-à-dire atomes, groupes d'atomes ou centres de réaction qui déterminent les propriétés physiques, chimiques ou pharmacologiques des médicaments.
L'analyse élémentaire est utilisée pour tester l'authenticité des substances médicinales organiques contenant des atomes de soufre, d'azote, de phosphore, d'halogènes, d'arsenic et de métaux dans la molécule. Les atomes de ces éléments se trouvent dans les composés organo-éléments médicinaux à l'état non ionisé ; une condition nécessaire pour tester leur authenticité est une minéralisation préalable.
Il peut s'agir de substances liquides, solides et gazeuses. Les composés gazeux et liquides ont principalement un effet narcotique. L'effet est réduit par F - Cl - Br - I. Les dérivés de l'iode ont principalement un effet antiseptique. Connexion C-F ; CI ; C-Br; C-Cl est covalent, donc pour l'analyse pharmaceutique, des réactions ioniques sont utilisées après minéralisation de la substance.
L'authenticité des préparations d'hydrocarbures halogénés liquides est déterminée par des constantes physiques (point d'ébullition, densité, solubilité) et la présence d'halogène. La manière la plus objective d’établir l’authenticité consiste à identifier les spectres IR du médicament et les échantillons standards.
Pour prouver la présence d'halogènes dans une molécule, le test de Beilstein et diverses méthodes de minéralisation sont utilisés.
Tableau 1. Propriétés des composés contenant des halogènes
|
Chloréthyl Aéthylii cloridum (DCI Ethylchlorure) |
Ftorotan 1,1,1-trifluoro-2chloro-2-bromoéthane (DCI Halothane) |
Bromcamphre 3-bromo-1,7,7,triméthylbicycloheptanone-2 |
|
|
Le liquide est transparent, incolore, facilement volatil, avec une odeur particulière, peu soluble dans l'eau et miscible à l'alcool et à l'éther dans n'importe quelle proportion. |
Le liquide est incolore, transparent, lourd, volatil, d'odeur caractéristique, légèrement soluble dans l'eau, miscible à l'alcool, à l'éther et au chloroforme. |
Poudre cristalline blanche ou cristaux incolores, odeur et goût, très peu soluble dans l'eau, facilement dans l'alcool et le chloroforme. |
|
|
Bilignostum pro injectionibus Bilignost Acide bis-(2,4,6-triiodo-3-carboxyanilide) adipique |
Bromé 2-bromoisovalérianyl-urée |
||
|
Poudre cristalline blanche, au goût légèrement amer, pratiquement insoluble dans l'eau, l'alcool, le chloroforme. |
Poudre cristalline blanche ou cristaux incolores à faible odeur spécifique, légèrement soluble dans l'eau, soluble dans l'alcool. |
essai de Beilstein
La présence d'halogène est prouvée par calcination de la substance à l'état solide sur un fil de cuivre. En présence d'halogènes, des halogénures de cuivre se forment, qui colorent la flamme en vert ou en bleu-vert.
Les halogènes d'une molécule organique sont reliés par une liaison covalente dont le degré de force dépend de la structure chimique du dérivé halogène ; par conséquent, diverses conditions sont nécessaires pour l'élimination d'un halogène et son transfert vers un état ionisé. Les ions halogénures résultants sont détectés par des réactions analytiques conventionnelles.
Chloroéthyle
· Méthode de minéralisation - ébullition avec une solution alcoolique d'alcali (étant donné le faible point d'ébullition, la détermination est effectuée avec un condenseur à reflux).
CH 3 CH 2 Cl + KOH c KCl + C 2 H 5 OH
L'ion chlorure résultant est détecté avec une solution de nitrate d'argent par la formation d'un précipité blanc de fromage.
Cl- + AgNO 3 > AgCl + NO 3 -
Ftorotan
· Méthode de minéralisation - fusion avec du sodium métallique
F 3 C-CHClBr + 5Na + 4H 2 O> 3NaF + NaCl + 2NaBr + 2CO 2
Les ions chlorure et bromure résultants sont détectés par une solution de nitrate d'argent par la formation de précipités blancs fromageux et jaunâtres.
L'ion fluorure est prouvé par des réactions :
Réaction avec une solution de rouge d'alizarine et une solution de nitrate de zirconium, en présence de F-, la couleur rouge vire au jaune clair ;
Interaction avec les sels de calcium solubles (un précipité blanc de formes de fluorure de calcium) ;
Réaction de décoloration du thiocyanate de fer (rouge).
· Lorsqu'il est ajouté au fluorothane conc. H 2 SO 4, le médicament est dans la couche inférieure.
Bromé
· Méthode de minéralisation - ébullition avec un alcali (hydrolyse alcaline en solution aqueuse), une odeur d'ammoniaque apparaît :
· Chauffage avec conc. acide sulfurique - l'odeur de l'acide isovalérique
Bromcamphre
· Méthode de minéralisation par la méthode de minéralisation par réduction (avec du zinc métallique en milieu alcalin)
L'ion bromure est déterminé par réaction avec la chloramine B.
Bilignost
· Méthode de minéralisation - chauffage à l'acide sulfurique concentré : on constate l'apparition de vapeurs violettes d'iode moléculaire.
· Spectroscopie IR - une solution à 0,001 % du médicament dans une solution d'hydroxyde de sodium 0,1 N dans la plage de 220 à 300 nm a un maximum d'absorption à l = 236 nm.
Iodoforme
Méthodes de minéralisation :
1) pyrolyse dans un tube à essai sec, de la vapeur d'iode violet est libérée
4CHI 3 + 5O 2 > 6I 2 + 4CO 2 + 2H 2 O
2) chauffage avec conc. acide sulfurique
2CHI 3 + H 2 SO 4 > 3I 2 + 2CO 2 + 2H 2 O + SO 3
Bonne qualité (pureté des hydrocarbures halogénés).
La qualité du chloréthyle et du fluorotane est vérifiée en établissant l'acidité ou l'alcalinité, l'absence ou la teneur acceptable en stabilisants (thymol dans le fluorotane - 0,01 %), les impuretés organiques étrangères, les impuretés de chlore libre (brome dans le fluorotane), les chlorures, les bromures et les non- résidu volatil.
1) Chloroéthyle : 1. Déterminer le point d’ébullition et la densité,
2. Impureté inacceptable de l'alcool éthylique (réaction de formation d'iodoforme)
2) Bilignost : 1. Chauffage avec kH 2 SO 4 et formation de vapeurs violettes I 2
2. Spectroscopie IR
3) Ftorotan : 1. Spectroscopie IR
2. point d'ébullition ; densité; indice de réfraction
3. il ne devrait y avoir aucune impureté Cl- et Br-
GF ne prévoit pas le dosage quantitatif du chloréthyle, mais il peut être réalisé par argentométrie ou mercurimétrie.
La méthode de dosage quantitatif est le titrage argentométrique inverse selon Volhard après minéralisation (pour la réaction, voir la définition de l'authenticité).
1. Réaction avant titrage :
titrage pharmaceutique et médicinal du chloroéthyle
NaBr + AgNO 3 > AgBrv + NaNO 3
2. Réaction de titrage :
AgNO 3 + NH 4 SCN > AgSCN v + NH 4 NO 3
3. Au point d'équivalence :
3NH 4 SCN + Fe(NH 4)(SO 4) 2 >
La méthode de détermination quantitative est le titrage argentométrique selon Kolthoff après minéralisation (pour les réactions, voir la définition de l'authenticité).
1. Réaction avant titrage :
3NH 4 SCN + Fe(NH 4)(SO 4) 2 > Fe (SCN) 3 + 2 (NH 4) 2 SO 4
quantité exacte rouge brunâtre
2. Réaction de titrage :
NaBr + AgNO 3 > AgBrv + NaNO 3
3. Au point d'équivalence :
AgNO 3 + NH 4 SCN > AgSCNv + NH 4 NO 3
blanchiment
Bilignost
La méthode de détermination quantitative est l'iodométrie indirecte après le clivage oxydatif du bilignost en iodate lorsqu'il est chauffé avec une solution de permanganate de potassium dans un milieu acide, l'excès de permanganate de potassium est éliminé à l'aide de nitrate de sodium et pour éliminer l'excès d'acide nitreux, une solution d'urée est ajoutée au mélange.
Titrant - solution de titsulfate de sodium 0,1 mol/l, indicateur - amidon, au point d'équivalence on observe la disparition de la couleur bleue de l'amidon.
Schéma de réaction :
t ; KMnO 4 +H 2 SO 4
RI 6 > 12 IO 3 -
Réaction de libération du substituant :
KIO 3 + 5KI + 3H 2 SO 4 >3I 2 + 3K 2 SO 4 + 3H 2 O
Réaction de titrage :
I 2 +2Na 2 S 2 O 3 > 2NaI+Na 2 S 4 O 6
Iodoforme
La méthode de détermination quantitative est le titrage argentométrique inverse selon Volhard après minéralisation.
Minéralisation:
CHI 3 + 3AgNO 3 + H 2 O> 3AgI + 3HNO 3 + CO 2
Réaction de titrage :
AgNO 3 + NH 4 SCN > AgSCN v + NH 4 NO 3
Au point d'équivalence :
3NH 4 SCN + Fe(NH 4)(SO 4) 2 > Fe (SCN) 3 v + 2 (NH 4) 2 SO 4
Stockage
Chloroéthyle en ampoules dans un endroit frais et sombre, ftorotan et bilignost en flacons en verre orange dans un endroit frais et sec, à l'abri de la lumière. Le bromcamphre est conservé dans des bouteilles en verre orange dans un endroit frais et sec.
Le chloréthyle est utilisé pour l'anesthésie locale, le fluorothane pour l'anesthésie. Le bromcamphre est utilisé comme sédatif (parfois pour arrêter la lactation). Bromizoval est un hypnotique ; le bilignost est utilisé comme agent de radiocontraste sous la forme d'un mélange de sels en solution.
Littérature
1. Pharmacopée d'État de l'URSS / Ministère de la Santé de l'URSS. - X éd. - M. : Médecine, 1968. - P. 78, 134, 141, 143, 186, 373 537
2. Pharmacopée d'État de l'URSS Vol. 1. Méthodes générales d'analyse. Matières premières végétales médicinales / Ministère de la Santé de l'URSS. - 11e éd., ajouter. - M. : Médecine, 1989. - S. 165-180, 194-199
3. Matériel de cours.
4. Chimie pharmaceutique. En 2 heures : manuel / V. G. Belikov - 4e éd., révisé. et supplémentaire - M. : MEDpress-inform, 2007. - P. 178-179, 329-332
5. Guide des cours de laboratoire en chimie pharmaceutique. Edité par A.P. Arzamastseva, pp.152-156.
Publié sur Allbest.ru
Annexe 1
Articles de pharmacopée
Bilignost
Acide bis-(2,4,6-triiodo-3-carboxyanilide) adipique
C 20 H 14 I 6 N 2 O 6 M. c. 1139.8
Description. Poudre finement cristalline blanche ou presque blanche au goût légèrement amer.
Solubilité. Pratiquement insoluble dans l'eau, l'alcool à 95 %, l'éther et le chloroforme, facilement soluble dans les solutions d'alcalis caustiques et d'ammoniaque.
Authenticité. Solution à 0,001% du médicament dans 0,1 N. une solution de soude caustique dans la région de 220 à 300 nm a un maximum d'absorption à une longueur d'onde d'environ 236 nm.
Lorsque 0,1 g du médicament est chauffé avec 1 ml d'acide sulfurique concentré, des vapeurs d'iode violet sont libérées.
Couleur de la solution. 2 g du médicament sont dissous dans 4 ml de 1 N. solution de soude, filtrer et laver le filtre à l'eau jusqu'à obtention de 10 ml de filtrat. La couleur de la solution obtenue ne doit pas être plus intense que la norme n° 4b ou n° 4c.
Testez avec du peroxyde d'hydrogène. A 1 ml de la solution obtenue, ajoutez 1 ml de peroxyde d'hydrogène ; aucun trouble ne devrait apparaître dans les 10 à 15 minutes.
Composés avec un groupe amino ouvert. 1 g du médicament est secoué avec 10 ml d'acide acétique glacial et filtré. A 5 ml de filtrat clair, ajoutez 3 gouttes de solution 0,1 mole de nitrite de sodium. Au bout de 5 minutes, la couleur qui apparaît ne doit pas être plus intense que le standard n°2g.
Acidité. 0,2 g du médicament est agité pendant 1 minute avec de l'eau bouillante (4 fois 2 ml chacune) et filtré jusqu'à l'obtention d'un filtrat clair. Je titre les filtrats combinés ! 0,05 n. solution d'hydroxyde de sodium (indicateur phénolphtaléine). Pas plus de 0,1 ml de 0,05 N ne doit être utilisé pour le titrage. solution de soude caustique.
Chlorures. Agiter 2 g du médicament avec 20 ml d'eau et filtrer jusqu'à l'obtention d'un filtrat clair. 5 ml de filtrat, portés à 10 ml avec de l'eau, doivent passer le test des chlorures (pas plus de 0,004 % dans la préparation).
Phosphore. 1 g du médicament est placé dans un creuset et réduit en cendres jusqu'à l'obtention d'un résidu blanc. 5 ml d'acide nitrique dilué sont ajoutés au résidu et évaporés à sec, après quoi le résidu dans le creuset est bien mélangé avec 2 ml d'eau chaude et filtré dans un tube à essai à travers un petit filtre. Le creuset et le filtre sont lavés avec 1 ml d'eau chaude en récupérant le filtrat dans le même tube à essai, puis ajouter 3 ml de solution de molybdate d'ammonium et laisser reposer 15 minutes dans un bain à une température de 38-40°. peut avoir une couleur jaunâtre, mais doit rester transparent (pas plus de 0,0001 % dans le médicament).
Monochlorure d'iode. Agiter 0,2 g du médicament avec 20 ml d'eau et filtrer jusqu'à l'obtention d'un filtrat clair. A 10 ml de filtrat, ajoutez 0,5 g d'iodure de potassium, 2 ml d'acide chlorhydrique et 1 ml de chloroforme. La couche de chloroforme doit rester incolore.
Fer. 0,5 g du médicament doit réussir le test de fer (pas plus de 0,02 % dans le médicament). La comparaison est effectuée avec un étalon préparé à partir de 3,5 ml de solution étalon B et 6,5 ml d'eau.
Les cendres de sulfate provenant de 1 g du médicament ne doivent pas dépasser 0,1 %.
Métaux lourds. Les cendres sulfatées provenant de 0,5 g du médicament doivent réussir le test de détection des métaux lourds (pas plus de 0,001 % dans le médicament).
Arsenic. 0,5 g du médicament doit réussir le test d'arsenic (pas plus de 0,0001 % dans le médicament).
Quantification. Environ 0,3 g de médicament (pesé exactement) est placé dans une fiole jaugée de 100 ml, dissous dans 5 ml de solution d'hydroxyde de sodium, ajouté avec de l'eau jusqu'au trait et mélangé. 10 ml de la solution obtenue sont placés dans un ballon d'une capacité de 250 ml, 5 ml d'une solution à 5% de permanganate de potassium sont ajoutés et soigneusement le long des parois du ballon, sous agitation, 10 ml d'acide sulfurique concentré, 0,5 -1 ml chacun, est ajouté et laissé 10 minutes. Ajoutez ensuite lentement, 1 goutte après 2-3 secondes, en remuant vigoureusement. solution de nitrite de sodium jusqu'à ce que le liquide se décolore et que le dioxyde de manganèse se dissolve. Après cela, ajoutez immédiatement 10 ml d'une solution d'urée à 10 % et remuez jusqu'à disparition complète des bulles, tout en éliminant le nitrite de sodium des parois du ballon. Ensuite, 100 ml d'eau, 10 ml d'une solution d'iodure de potassium fraîchement préparée sont ajoutés à la solution et l'iode libéré est titré avec 0,1 N. solution de thiosulfate de sodium (indicateur - amidon).
1 ml 0,1 n. La solution de thiosulfate de sodium correspond à 0,003166 g de C 20 H 14 l 6 N 2 0 6, qui doit être d'au moins 99,0 % dans la préparation.
Stockage. Liste B. Dans des bocaux en verre orange, à l'abri de la lumière.
Agent de contraste pour rayons X.
Iodoforme
Triiodométhane
СНI 3 М.в. 393,73
Description. Petits cristaux lamellaires brillants ou fine poudre cristalline de couleur jaune citron, odeur persistante caractéristique piquante. Volatil déjà aux températures ordinaires, il est distillé avec de la vapeur d'eau. Les solutions du médicament se décomposent rapidement sous l'influence de la lumière et de l'air, libérant de l'iode.
Solubilité. Pratiquement insoluble dans l'eau, peu soluble dans l'alcool, soluble dans l'éther et le chloroforme, légèrement soluble dans le glycérol. huiles grasses et essentielles.
Authenticité, 0,1 g du médicament est chauffé dans un tube à essai sur la flamme d'un brûleur ; de la vapeur d'iode violette est libérée.
Point de fusion 116--120° (avec décomposition).
Colorants. 5 g du médicament sont agités vigoureusement pendant 1 minute avec 50 ml d'eau et filtrés. Le filtrat doit être incolore.
Acidité ou alcalinité. A 10 ml de filtrat, ajoutez 2 gouttes de solution de bleu de bromothymol. La couleur jaune-vert qui apparaît doit virer au bleu avec l'ajout de 0,1 ml maximum de 0,1 N. solution de soude caustique ou jaune en ajoutant au maximum 0,05 ml de 0,1 N. solution d'acide chlorhydrique.
Halogènes. 5 ml du même filtrat, dilué avec de l'eau à 10 ml, doivent passer le test des chlorures (pas plus de 0,004% dans la préparation).
Sulfates. 10 ml du même filtrat doivent réussir le test des sulfates (pas plus de 0,01% dans la préparation).
Les cendres provenant de 0,5 g du médicament ne doivent pas dépasser 0,1 %.
Quantification. Environ 0,2 g du médicament (pesé exactement) est placé dans une fiole conique d'une capacité de 250 à 300 ml, dissous dans de l'alcool à 25 ou 95 %, 25 ml de 0,1 N sont ajoutés. solution de nitrate d'argent, 10 ml d'acide nitrique et porter à reflux au bain-marie pendant 30 minutes en protégeant le ballon de réaction de la lumière. Le réfrigérateur est lavé à l'eau, 100 ml d'eau sont ajoutés dans le ballon et l'excès de nitrate d'argent est titré avec 0,1 N. solution de thiocyanate d'ammonium (indicateur - alun d'ammonium ferrique).
Parallèlement, une expérience de contrôle est réalisée.
1 ml 0,1 n. la solution de nitrate d'argent correspond à 0,01312 g de CHI 3, qui doit être au minimum à 99,0% dans la préparation.
Stockage. Dans un récipient bien fermé, à l'abri de la lumière, dans un endroit frais.
Publié sur Allbest.ru
Documents similaires
Le concept de réfraction comme mesure de la polarisabilité électronique des atomes, des molécules et des ions. Évaluation de l'indice de réfraction pour l'identification de composés organiques, de minéraux et de substances médicinales, leurs paramètres chimiques, analyse quantitative et structurelle.
travail de cours, ajouté le 05/06/2011
Opérations de base lorsqu'on travaille dans un laboratoire de chimie organique. Les constantes physiques les plus importantes. Méthodes de détermination de la structure des composés organiques. Fondamentaux de structure, propriétés et identification des composés organiques. Synthèses de composés organiques.
manuel de formation, ajouté le 24/06/2015
Etude des fondements théoriques des méthodes de précipitation des substances médicinales organiques et inorganiques. Analyse des caractéristiques de l'interaction des substances médicinales avec des indicateurs dans les méthodes de précipitation. Méthodes indicatives pour déterminer le point final du titrage.
travail de cours, ajouté le 30/01/2014
Dimérisation oxydative du méthane. Mécanisme d'activation catalytique du méthane. Préparation de composés organiques par méthylation oxydative. Transformations oxydatives de composés organiques contenant un groupe méthyle en présence d'un catalyseur.
thèse, ajoutée le 11/10/2013
Prise en compte de réactions basées sur la formation de composés métalliques complexes et sans leur participation. Le concept de groupes fonctionnels-analytiques et analytiques-actifs. Utilisation de composés organiques comme indicateurs de méthodes titrimétriques.
travail de cours, ajouté le 01/04/2010
Structure chimique - la séquence de connexions d'atomes dans une molécule, l'ordre de leur interconnexion et leur influence mutuelle. La connexion des atomes qui composent les composés organiques ; dépendance des propriétés des substances sur le type d'atomes, leur quantité et leur ordre d'alternance.
présentation, ajouté le 12/12/2010
L'isomérie est le phénomène d'existence de composés de composition identique, mais de structure et de propriétés différentes. Isomérie interclasse déterminé par la nature du groupe fonctionnel. Types d'isomérie spatiale. Types de nomenclature des composés organiques.
présentation, ajouté le 12/03/2017
Méthodes de base pour prédire les enthalpies de formation de composés organiques : méthodes de mécanique moléculaire et méthodes additives. Méthode Benson et méthode Tatevsky. Alkylbenzènes et leurs dérivés fonctionnels : halobenzènes, polyphényles, pyridines.
travail de cours, ajouté le 17/01/2009
Halogénation des composés aromatiques : mécanisme du processus. Calcul d'indicateurs de mono- et dichloration de composés organiques. Consommation de réactifs avec un rendement maximal du produit cible dans des réactions complexes. Sélection d'un mécanisme de réaction approprié.
résumé, ajouté le 15/02/2012
La vie comme un processus physique et chimique continu. Caractéristiques générales des composés naturels. Classification des composés naturels de faible poids moléculaire. Critères de base pour la classification des composés organiques. Types et propriétés des liaisons, influence mutuelle des atomes.