Die wichtigsten Arten von Kristallstrukturen. Der Wert des Wissens über die Kristallstruktur von Metallen
Lesen Sie auch
Je nach Art der Struktureinheiten eines Stoffes werden Rahmen- (atomare), metallische, ionische und molekulare Strukturen unterschieden. Es gibt auch kombinierte Arten von Strukturen.
BEI rahmen Struktur sind die Atome eines oder mehrerer chemischer Elemente durch kovalente chemische Bindungen verbunden. Als Ergebnis wird die Wahl einer bestimmten Struktur durch die Orientierung der Verknüpfungen diktiert. Es gibt keine isolierten Atomgruppen in der Struktur; ein Netzwerk aus kovalenten Bindungen bedeckt die gesamte Struktur. Das bekannteste Beispiel für eine Substanz mit Skelettstruktur ist Diamant. Die Elementarzelle des Diamanten ist in Abb. 1 dargestellt. 8.7. Kohlenstoffatome befinden sich an den Ecken einer kubischen Zelle, den Zentren aller Flächen, und besetzen in einem Schachbrettmuster die Zentren von vier der acht Würfel, in die eine Einheitszelle unterteilt werden kann. Von diesen Atomen innerhalb der Zelle sind die kovalenten Bindungen tetraedrisch zum Kohlenstoffatom an einem der Eckpunkte und zu drei Kohlenstoffatomen an den Flächen gerichtet. Die Abstände zwischen allen Kohlenstoffatomen betragen 154 pm. Viele Substanzen haben diamantartige Strukturen. Darunter sind Silizium, Siliziumcarbid SiC, Zinksulfid (Zinkblende) ZnS. In dieser Substanz befinden sich Zinkatome an den Ecken und an den Flächen der Einheitszelle, und Schwefelatome besetzen Plätze innerhalb der Zelle. Somit ist die Struktur dieser Substanz, die traditionell als Salze bezeichnet wird, nicht ionisch, sondern gerüstartig.
Ein Kristall einer Substanz mit einer Gerüststruktur kann als einzelnes Molekül betrachtet werden. Solche Substanzen sind thermisch stabil, in Wasser praktisch unlöslich, haben hohe Schmelzpunkte und eine hohe Härte.
Metall die Struktur unterscheidet sich von der Rahmenstruktur dadurch, dass die Anordnung der Atome nicht durch die Richtung der Bindungen bestimmt wird, sondern nur durch den Zustand der dichtesten Packung von Atomkugeln. Für die meisten Metalle sind nur drei Arten von Einheitszellen charakteristisch – kubisch raumzentriert, kubisch flächenzentriert und hexagonal kompakt. Viele Metalle zeigen Polymorphismus, Veränderung der Kristallstruktur beim Erhitzen.
Reis. 8.7.
Keile zeigen Bindungen zwischen Kohlenstoffatomen innerhalb einer Zelle
Ionisch Die Struktur ist aus alternierenden Ionen mit entgegengesetzten Ladungen aufgebaut. Natriumchlorid hat eine solche Struktur (siehe Abb. 2.8). Die Positionen der Natrium- und Chloridionen sind vollständig austauschbar. Chlorionen können an den Ecken der Zelle und in den Mitten der Flächen platziert werden. Dann befinden sich die Natriumionen in der Mitte der Rippen und im Zentrum der Zelle. Sie können das Gegenteil tun, d.h. alle Ionen tauschen. Eine solche Struktur kann man sich als zwei flächenzentrierte Gitter - eines mit Na + -Ionen und das andere mit C1~ -Ionen - vorstellen, die mit einer Verschiebung um die halbe Würfelkantenlänge ineinander geschoben sind.
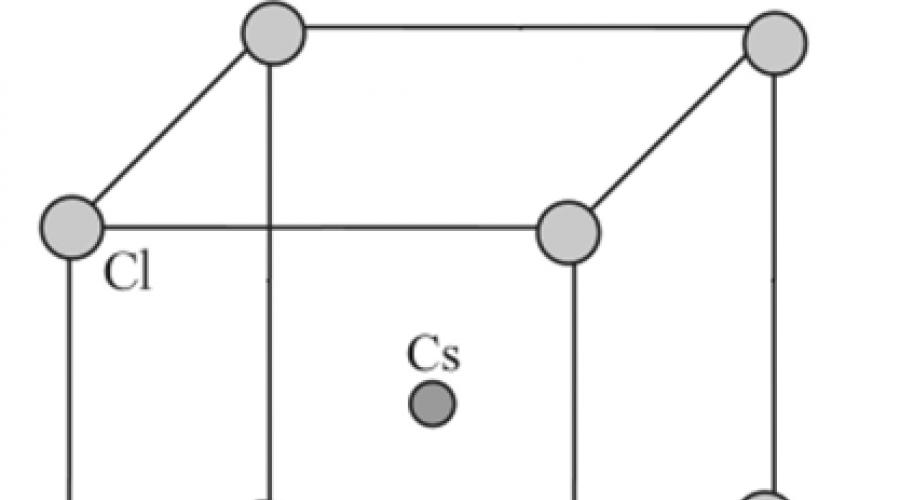
Das Aussehen der einen oder anderen Ionenstruktur hängt hauptsächlich von den Ladungen der Ionen und dem Verhältnis ihrer Radien ab. In Cäsiumchlorid, einem schwereren Alkalimetall als Natrium, wird der Radius des Kations deutlich vergrößert, wodurch seine Koordinationszahl auf acht ansteigt. In einer kubischen Zelle ist jedes Cäsiumion von acht Chloridionen umgeben (Abb. 8.8). Diese Struktur kann auch als zwei kubische Gitter dargestellt werden, die aus Cäsium-Ionen und Chlor-Ionen bestehen, die ineinander eingefügt sind, so dass sich ein Ion einer Art im Zentrum einer Zelle mit Ionen einer anderen Art befindet.

Reis. 8.8.
Substanzen mit ionischer Struktur zeichnen sich durch hohe Schmelzpunkte aufgrund der erheblichen Energie der elektrostatischen Anziehung von Ionen aus. Viele ionische Substanzen sind gut wasserlöslich.
Substanzen mit molekular Strukturen unterscheiden sich stark von den oben betrachteten durch niedrige Schmelzpunkte. Darunter sind Flüssigkeiten und Gase. Röntgenbeugungsstudien solcher Substanzen zeigen kurze interatomare Abstände innerhalb von Molekülen und deutlich verlängerte Abstände zwischen denselben Atomen in verschiedenen Molekülen. Beispielsweise beträgt in Jod-12-Kristallen (Abb. 8.9) der Abstand zwischen Atomen in einem Molekül 272 pm, der Abstand zwischen Molekülen in einer Schicht 350 nm und der nächste Abstand zwischen Atomen in verschiedenen Schichten 397 pm.

Reis. 8.9.
Substanzen, die aus mehratomigen Molekülen bestehen, bilden sehr komplexe Strukturen. Ohne eine Röntgenuntersuchung wäre es einfach unmöglich, ihre Struktur zu verstehen. Wir erinnern uns an die DNA-Moleküle mit einer Doppelhelixstruktur. Die Bestimmung ihrer Struktur eröffnete eine neue Etappe in der Entwicklung der Biologie.
Es ist offensichtlich, dass ein Molekül nicht in einem Knoten der Kristallstruktur lokalisiert werden kann, da es sich um eine bestimmte Menge von Atomen handelt. Auf Abb. 8.10 ist als Beispiel die Struktur der Komplexverbindung |Pt (CN) 2 (NH 3) (NH 2 CH 3) | Die Elementarzelle wird als Projektion entlang der Achse dargestellt U. Die Ecken der Zelle sind nicht mit Atomen besetzt. Die planaren Moleküle der Komplexverbindung sind in der Projektion von der Seite zu sehen. Die gepunktete Linie zeigt Wasserstoffbrückenbindungen zwischen Ammoniakmolekülen in verschiedenen Molekülen der Komplexverbindung. Symmetrieachsen zweiter Ordnung verlaufen parallel zur Achse U. Einer von ihnen geht durch die Mitte der Zelle. Acht Moleküle in einer Elementarzelle befinden sich auf zwei Ebenen entlang der Achse Bei im Schachbrettmuster. Dieses Beispiel gibt eine Vorstellung von der Komplexität molekularer Strukturen.

Reis. 8.10.Projektion der Einheitszelle einer komplexen Verbindung entlang der AchseY
1.4. Haupttypen von Kristallstrukturen
Die Punktanordnung von Atomen in räumlichen Gittern ist vereinfacht und für die Untersuchung von Kristallstrukturen ungeeignet, wenn der Abstand zwischen den nächsten Atomen oder Ionen bestimmt wird. Die physikalischen Eigenschaften kristalliner Strukturen hängen jedoch von der chemischen Natur der Substanzen, der Größe der Atome (Ionen) und den Wechselwirkungskräften zwischen ihnen ab. Daher werden wir in Zukunft davon ausgehen, dass Atome oder Ionen die Form einer Kugel haben und sich dadurch auszeichnen Wirkradius, wobei man darunter den Radius ihrer Einflusssphäre versteht, der gleich der Hälfte des Abstands zwischen den beiden nächsten benachbarten Atomen oder Ionen derselben Art ist. In einem kubischen Gitter ist der effektive Atomradius a 0 /2.
Der effektive Radius hat in jeder einzelnen Struktur unterschiedliche Eigenwerte und hängt von der Art und Anzahl der Nachbaratome ab. Die Atomradien verschiedener Elemente lassen sich nur vergleichen, wenn sie Kristalle mit gleicher Koordinationszahl bilden. Koordinierungsnummer z eines gegebenen Atoms (Ions) ist die Anzahl der nächsten ähnlichen Atome (Ionen), die es in der Kristallstruktur umgeben. Wenn wir die Zentren benachbarter Teilchen gedanklich mit geraden Linien miteinander verbinden, erhalten wir
Koordinationspolyeder; in diesem Fall befindet sich das Atom (Ion), für das ein solcher Polyeder konstruiert ist, in seinem Zentrum.
Die Koordinationszahl und das Verhältnis der effektiven Partikelradien stehen in einem bestimmten Zusammenhang: Je kleiner der Partikelgrößenunterschied, desto größer z.
Je nach Kristallstruktur (Gittertyp) kann z von 3 bis 12 variieren. Wie unten gezeigt wird, ist in der Struktur von Diamant z = 4, in Steinsalz z = 6 (jedes Natriumion ist von sechs Chloridionen umgeben) . Für Metalle ist die Koordinationszahl z = 12 typisch, für kristalline Halbleiter z = 4 oder z = 6. Für Flüssigkeiten wird die Koordinationszahl statistisch als durchschnittliche Anzahl der nächsten Nachbarn eines beliebigen Atoms bestimmt.
Die Koordinationszahl hängt mit der Packungsdichte der Atome in der Kristallstruktur zusammen. Relative Packungsdichte–
es ist das Verhältnis des von den Atomen eingenommenen Volumens zum Gesamtvolumen der Struktur. Je höher die Koordinationszahl, desto höher die relative Packungsdichte.

Abschnitt 1. Grundlagen der physikalisch-chemischen Kristallographie
Das Kristallgitter neigt dazu, ein Minimum an freier Energie zu haben. Dies ist nur möglich, wenn jedes Teilchen mit möglichst vielen anderen Teilchen wechselwirkt. Mit anderen Worten, die Koordinationszahl sollte maximal m sein Die Tendenz zur dichten Packung ist charakteristisch für alle Arten von Kristallstrukturen.
Stellen Sie sich eine planare Struktur vor, die aus Atomen der gleichen Art besteht, die sich berühren und den größten Teil des Raums ausfüllen. In diesem Fall ist nur eine Art der dichtesten Packung benachbarter Atome möglich: um das Zentrum herum
die Schwerpunkte fallen auf die Hohlräume der ersten Schicht. Dies ist im rechten Bild in Abb. 1.10, a (Draufsicht), wo die Projektionen der Atome der zweiten Schicht hellgrau gemalt sind. Die Atome der zweiten Schicht bilden ein Grunddreieck (gezeigt durch eine durchgezogene Linie), wobei die Spitze nach oben zeigt.
Reis. 1.10. Die Schichtenfolge beim Packen von Kugeln gleicher Größe in Strukturen zweier Typen: (a) ABAB... mit hexagonal dichter Packung (HCP); b - ABSABC ... mit dem dichtesten kubischen Paket (K PU), was ein flächenzentriertes kubisches (fcc) Gitter ergibt. Zur Verdeutlichung sind die dritte und vierte Schicht unvollständig gefüllt dargestellt.

Kapitel 1. Elemente der Kristallphysik
Die Anordnung der Atome der dritten Schicht kann auf zwei Arten erfolgen. Wenn die Schwerpunkte der Atome der dritten Schicht über den Schwerpunkten der Atome der ersten Schicht liegen, wird das Verlegen der ersten Schicht wiederholt (Abb. 1.10, a). Die resultierende Struktur ist hexagonal dichte Packung(GPU). Es kann als Folge von Schichten ABABABAB ... in Richtung der Z-Achse dargestellt werden.
Befinden sich die Atome der dritten Schicht C (in Abb. 1.10, b rechts dunkelgrau dargestellt) über anderen Hohlräumen der ersten Schicht und bilden ein gegenüber Schicht B um 180° gedrehtes Grunddreieck (gestrichelt dargestellt) , und die vierte Schicht identisch mit der ersten ist, stellt dann die resultierende Struktur dar kubisch dichteste Packung(FCC), was einer kubisch-flächenzentrierten Struktur (FCC) mit einer Schichtenfolge ABSABCABSABC ... in Richtung der Z-Achse entspricht.
Für die dichtesten Packungen ist z = 12. Deutlich wird dies am Beispiel der zentralen Kugel in Schicht B: Ihre nächste Umgebung besteht aus sechs Kugeln in Schicht A und drei Kugeln darunter und darüber in Schicht B
(Abb. 1.10, a).
Verschiedene Strukturen werden neben der Koordinationszahl z auch durch die Packungsdichte charakterisiert, die als Verhältnis des von Atomen eingenommenen Volumens V at zum Volumen der gesamten Bravais-Zelle V Zelle eingeführt wird. Atome werden durch feste Kugeln mit Radius r dargestellt, daher V at = n (4π/3)r 3, wobei n die Anzahl der Atome in einer Zelle ist.
Das Volumen der kubischen Zelle V-Zelle \u003d a 0 3, wobei a 0 die Gitterperiode ist. Für eine HCP-Zelle mit sechseckiger Grundfläche ist S = 3a 0 2 2 3
und Höhe c = 2a 0 23 erhalten wir V cell = 3a 0 3 2 .
Die entsprechenden Parameter der Kristallstrukturen - primitiv kubisch (PC), kubisch raumzentriert (BCC), kubisch flächenzentriert (FCC), hexagonal dicht gepackt (HCP) - sind in der Tabelle angegeben. 1.2. Die Atomradien werden unter Berücksichtigung dessen geschrieben, dass sie sich entlang der Kanten des Würfels in der PC-Struktur (2r = a 0 ), entlang der räumlichen Diagonalen (4r = a 0 3) in der bcc-Struktur und entlang der Diagonalen der Gesichter (4r = a 0 2)
in der fcc-Struktur.
So ist in den dichtesten gepackten Strukturen (fcc und hcp) mit z = 12 das Zellvolumen zu 74 % mit Atomen besetzt. Wenn die Koordinationszahl auf 8 und 6 abnimmt, nimmt die Packungsdichte auf 68 (bcc) bzw. 52 % (PC) ab.
![]()
Tabelle 1.2
Parameter von kubischen und hexagonalen Kristallen
Kristallparameter |
|||||
Koordinierungsnummer z |
|||||
Anzahl der Atome n in einer Zelle |
|||||
Atomradius r |
ein 0 /2 |
eine 2 4 |
ein 0 /2 |
||
Das Volumen eines Atoms, V at / n |
|||||
ein 0 3 π 6 |
a3π |
a 3 π 2 24 |
π ein 0 3 6 |
||
Packungsdichte, |
|||||
π 3 8 \u003d 0,6 |
π 2 6 \u003d 0,74 |
π 2 6 \u003d 0,74 |
|||
V bei / V-Zelle |
|||||
Es wurde bereits festgestellt, dass das System während der Kristallisation einer Substanz dazu neigt, ein Minimum an freier Energie bereitzustellen. Einer der Faktoren, die die potentielle Wechselwirkungsenergie zwischen Partikeln reduzieren, ist ihre maximale Annäherung und die Herstellung einer gegenseitigen Verbindung mit der größtmöglichen Anzahl von Partikeln, d. h. der Wunsch nach einer dichteren Packung mit der größtmöglichen Koordinationszahl.
Die Tendenz zur dichtesten Packung ist charakteristisch für alle Arten von Strukturen, am ausgeprägtesten jedoch bei Metall-, Ionen- und Molekülkristallen. Bei ihnen sind die Bindungen ungerichtet oder schwach gerichtet (s. Kap. 2), also für Atome Ionen
und Molekülen ist das Modell fester inkompressibler Kugeln durchaus akzeptabel.
Die in Abb. 1 gezeigten Bravais-Translationsgitter. 1.3
und im Tisch. 1.1 sind nicht alle Möglichkeiten zum Aufbau von Kristallstrukturen erschöpft, vor allem für chemische Verbindungen. Der Punkt ist, dass die periodische Wiederholung der Bravais-Zelle ein Translationsgitter ergibt, das nur aus gleichartigen Teilchen (Molekülen, Atomen, Ionen) besteht. Daher kann die Struktur einer komplexen Verbindung durch eine Kombination von Bravais-Gittern aufgebaut werden, die auf bestimmte Weise ineinander eingefügt werden. Halbleiterkristalle nutzen also eine gerichtete kovalente (unpolare oder polare) Bindung, die meist durch eine Kombination von mindestens zwei Gittern realisiert wird, die einzeln recht dicht gepackt sind, aber letztlich kleine Koordinationszahlen des „gesamten“ Gitters (bis zu z = 4).
Es gibt Stoffgruppen, die sich durch eine identische räumliche Anordnung der Atome auszeichnen und sich nur in den Parametern (aber nicht in der Art) des Kristallgitters voneinander unterscheiden.

Daher kann ihre Struktur mit einem einzigen räumlichen Modell beschrieben werden ( ein Strukturtyp), die die spezifischen Werte der Gitterparameter für jede Substanz angibt. Somit gehören Kristalle verschiedener Substanzen zu einer begrenzten Anzahl von Strukturtypen.
Die häufigsten Arten von Strukturen sind:
in Metallkristallen:
Struktur von Wolfram (OC-Gitter); Kupferstruktur (fcc-Gitter), Magnesiumstruktur (hcp-Gitter);
in dielektrischen Kristallen:
Struktur von Natriumchlorid (doppeltes HCC-Gitter); Struktur von Cäsiumchlorid (doppeltes PC-Gitter);
in Halbleiterkristallen:
Diamantstruktur (doppeltes fcc-Gitter); Sphaleritstruktur (doppeltes GCC-Gitter); Wurtzitstruktur (Doppel-HP-U-Gitter).
Betrachten wir kurz die Eigenschaften und Realisierbarkeit der oben aufgeführten Strukturen und der ihnen entsprechenden Bravais-Gitter.
1.4.1. Metallische Kristalle
Struktur von Wolfram(Abb. 1.1 1, aber). Das kubisch-raumzentrierte Gitter ist keine dichteste Struktur, es hat eine relative Packungsdichte von 0,6 8 und eine Koordinationszahl z = 8. Die (11 1)-Ebenen sind am dichtesten gepackt.
Reis. 1.11. Arten von kubischen Gittern: (a) kubisch raumzentriert (BCC); b - einfache Kubik

Abschnitt 1. Grundlagen der physikalisch-chemischen Kristallographie
Neben Wolfram W haben alle Alkali- und Erdalkalimetalle sowie die meisten Refraktärmetalle ein bcc-Gitter: Chrom Cr, Eisen Fe, Molybdän Mo, Zirkon Zr, Tantal Ta, Niob Nb usw. Letzteres findet Folgendes Erläuterung. In der bcc-Zelle für das Zentralatom sind die nächsten Nachbarn die Atome an den Eckpunkten des Würfels (z = 8). Sie sind auf Abstand voneinander
sechs zentrale Atome in benachbarten Zellen (zweite Koordinationssphäre), was die Koordinationszahl praktisch auf z 14 erhöht. Dies ergibt einen Gesamtenergiegewinn, der den negativen Beitrag einer geringfügigen Erhöhung der durchschnittlichen Abstände zwischen Atomen im Vergleich zum fcc-Gitter kompensiert, wobei die Atome einen Abstand von d = a 0 ( 2) 2 = 0,707a 0 haben. Dadurch ist die
Kristallisation, die sich in ihrem hohen Schmelzpunkt manifestiert, der für Wolfram 3422 ºС erreicht. Zum Vergleich: Eine einfache kubische Struktur (Abb. 1.11, b) mit z = 8 hat eine lockere Packung und kommt nur in Polonium vor.
Die in Abb. 1 gezeigte Kupferstruktur (fcc-Gitter). 1,12, a, bezieht sich auf dicht gepackte Strukturen, hat eine relative Packungsdichte von 0,74 und eine Koordinationszahl z = 12. Neben Kupfer Cu ist es charakteristisch für viele Metalle wie Gold Au, Silber Ag, Platin Pt, Nickel Ni, Aluminium Al, Blei Pb, Palladium Pd, Thorium Th usw.
Reis. 1.12. Strukturen dicht gepackter Kristallgitter: a – kubisch flächenzentriert (Kupferstruktur); b - hexagonal dicht gepackt (Magnesiumstruktur)

Kapitel 1. Elemente der Kristallphysik
Diese Metalle sind relativ weich und duktil. Der Punkt ist, dass in kupferartigen Strukturen die tetraedrischen und oktaedrischen Hohlräume im fcc-Gitter nicht mit anderen Partikeln gefüllt sind. Dies ermöglicht aufgrund der Nichtrichtung von Bindungen zwischen Atomen ihre Verschiebung entlang der sogenannten gleitende Ebenen. Im fcc-Gitter sind dies die Ebenen maximaler Packung (111), von denen eine in Abb. 1.12, a.
Struktur von Magnesium(hcp-Gitter) in Abb. 1.12, b, ist nicht nur für Magnesium Mg charakteristisch, sondern auch für Cadmium Cd, Zink Zn, Titan Ti, Thallium Tl, Beryllium Be usw. sowie für die meisten Seltenerdelemente. Im Gegensatz zum PC-Gitter ist das hcp-Gitter in Abb. 1.12, b hat eine Schicht B (schraffiert), die sich in der Mitte zwischen den Basisschichten A in einem festen Abstand befindet
mit 2 = a 0 2 3 (mit einer beobachteten Abweichung von bis zu 10 % für einige
andere Metalle). Die Atome in den Schichten B sind dicht gepackt über den Mittelpunkten der Dreiecke in der Basisebene (0001) platziert.
1.4.2. Dielektrische Kristalle
Struktur von Natriumchlorid(Abb. 1.13, aber) beschrieben werden
san als zwei kubisch flächenzentrierte Gitter (Strukturtyp Kupfer), die um eine halbe Gitterperiode (a 0 /2) entlang einer der Kanten verschoben sind<100>.
Große Chloranionen Cl– besetzen die Plätze der fcc-Zelle und bilden eine kubisch dichteste Packung, in der Natriumkationen Na+ mit geringerer Größe nur oktaedrische Lücken füllen. Mit anderen Worten, in der NaCl-Struktur ist jedes Kation von vier Anionen in der (100)-Ebene und zwei Ionen in der senkrechten Ebene umgeben, die den gleichen Abstand vom Kation haben. Als Ergebnis findet eine oktaedrische Koordination statt. Dies gilt gleichermaßen für Anionen. Daher ist das Verhältnis der Koordinationszahlen von Untergittern 6:6.
Struktur von Cäsiumchlorid CsCl (Doppel-PC-Gitter),
in Abb. gezeigt. 1.13, b, besteht aus zwei primitiven kubischen Gittern, die um die halbe Volumendiagonale verschoben sind. Tatsache ist, dass Cäsiumionen größer sind als Natriumionen und nicht in die oktaedrischen (und noch mehr in die tetraedrischen) Lücken des Chlorgitters passen, wenn es vom fcc-Typ wäre, wie in der Struktur von NaCl. In der CsCl-Struktur ist jedes Cäsium-Ion von acht Chlorid-Ionen umgeben und umgekehrt.
Auch andere Halogenide kristallisieren zu solchen Strukturen, beispielsweise Cs (Br, I), Rb (Br, I), Tl (Br, Cl), Halbleiterverbindungen vom AIV-BVI-Typ und viele Legierungen von Seltenerdelementen. Ähnliche Strukturen werden auch in heteropolaren ionischen Verbindungen beobachtet.
1.4.3. Halbleiterkristalle
Struktur eines Diamanten ist eine Kombination zweier FCC-Gitter, die ineinander gesteckt und entlang der Raumdiagonale um ein Viertel der Länge verschoben sind (Abb. 1.14, a). Jedes Atom ist von vier umgeben, die sich an den Ecken des Tetraeders befinden (dicke Linien in Abb. 1.14, a). Alle Bindungen in der Diamantstruktur sind gleich, entlang gerichtet<111>und bilden Winkel von 109º 28 " zueinander. Das Diamantgitter gehört zu locker gepackten Strukturen mit einer Koordinationszahl z = 4. Germanium, Silizium, graues Zinn kristallisieren in der Diamantstruktur. Neben Diamant kristallisieren darin auch elementare Halbleiter Art der Struktur - Silizium Si, Germanium Ge , zinngraues Sn.
Struktur von Sphalerit(doppeltes fcc-Gitter). Wenn zwei kubisch flächenzentrierte Hilfsgitter von verschiedenen Atomen gebildet werden, entsteht eine neue Struktur, die als ZnS-Sphaleritstruktur oder bezeichnet wird Zinkblende(Abb. 1.14, b).

Kapitel 1. Elemente der Kristallphysik
Reis. 1.14. Strukturen von Diamant (a), Falerit (b) und Wurtzit (c). Fette Linien zeigen t-Tetraederbindungen
Viele Halbleiterverbindungen vom Typ AIII BV (Galliumarsenid GaAs, Galliumphosphid GaP, Indiumphosphid InP, Indiumantimonid I nSb usw.) und Typ AII BVI (Zinkselenid ZnSe, Tellurzink ZnTe, Cadmiumsulfid CdS, Selenid Cadmium
Die Struktur von Sphalerit ist identisch mit der Struktur von Diamant mit einer tetraedrischen Umgebung von Atomen (Abb. 1.14, a), nur ein fcc-Untergitter ist von Gallium-Ga-Atomen und das andere von Arsen-As-Atomen besetzt. In der GaAs-Zelle gibt es kein Symmetriezentrum, d. h. die Struktur ist in vier Richtungen m polar< 111 >. Zwischen dicht gepackten 111)- und (111)-Ebenen wird ein Unterschied beobachtet: Wenn eine von ihnen Ga-Atome enthält, enthält die andere As-Atome. Dies bewirkt die Anisotropie der Oberflächeneigenschaften (Mikrohärte, Adsorption, chemisches Ätzen etc.).
In der Sphaleritstruktur sind die dreieckigen Basen der Tetraeder jeder Schicht in der gleichen Weise orientiert wie die Basen der Tetraeder der vorherigen Schicht.
Struktur von Wurtzit(Doppel-HCP-Gitter) in Abb. 1.14, c, ist charakteristisch für die hexagonale Modifikation von Zinksulfid. ZnS-ähnliche Halbleiter wie Cadmiumsulfid CdS und Cadmiumselenid CdSe haben eine solche Struktur. Die meisten AII-B-VI-Verbindungen sind durch den „Sphalerit-Wurtzit“-Phasenübergang gekennzeichnet. Die Wurtzitstruktur wird realisiert, wenn das Nichtmetallatom kleine Abmessungen und eine hohe Elektronegativität aufweist.
Auf Abb. Abbildung 1.14c zeigt eine primitive Wurtzit-Zelle für ZnS in Form eines geraden Prismas mit einer Raute an der Basis und einem Winkel von 120° in der Mitte eines Sechsecks, das aus drei solchen Prismen gebildet wird (von denen zwei in der Abbildung dargestellt sind). .
Reis. 17. Schneeflocken – Skeletteiskristalle
Aus Erfahrung ist bekannt, dass in einer kristallinen Substanz die physikalischen Eigenschaften in parallelen Richtungen gleich sind, und die Vorstellung von der Struktur von Substanzen erfordert, dass die Teilchen (Moleküle, Atome oder Ionen), aus denen der Kristall besteht, von einem entfernt sind eine andere in bestimmten endlichen Entfernungen. Basierend auf diesen Annahmen ist es möglich, ein geometrisches Diagramm der Kristallstruktur zu erstellen. Dazu kann die Position jedes Bestandteilspartikels mit einem Punkt markiert werden. Alles kristallinDas Gebäude wird dann als ein System von Punkten präsentiert, die regelmäßig im Raum und für jede Parallele angeordnet sindRichtungen des Abstands zwischen den Punkten sind gleich. Eine solche korrekte Anordnung von Punkten im Raum heißt
räumliches Gitter, und wenn jeder Punkt die Position eines Atoms, Ions oder Moleküls in einem Kristall darstellt - ein Kristallgitter.
Den Aufbau eines räumlichen Gitters kann man sich wie folgt vorstellen.
Eine 0(Abb. 18) bezeichnet das Zentrum eines Atoms oder Ions. Das gleiche Zentrum, das ihm am nächsten ist, sei mit dem Punkt A bezeichnet, dann, auf der Grundlage der Homogenität des Kristalls, in einem Abstand EIN 1 EIN 2 \u003d EIN 0 EIN 1 muss das Zentrum sein A2; Wenn wir dieses Argument weiter fortsetzen, können wir eine Reihe von Punkten erhalten: A 0, A 1, A 2, A 3 ...
Nehmen wir an, dass der nächstgelegene Punkt zu Eine 0 in die andere Richtung wird R0, dann muss es ein Teilchen geben S0 auf Distanz R 0 S 0= L 0 R 0 usw., d. h. es wird eine weitere Reihe identischer Punkte erhalten A 0 , R 0 , S 0… Wenn durch R 0 , S 0 usw. Linien parallel zu A 0, A 1, A 2 ziehen, erhalten Sie die gleichen Reihen R 0 , R 1 , R 2 , S 0 , S 1 , S 2 ... usw

Reis. 18. Räumliches Gitter
Als Ergebnis der Konstruktion wurde ein Gitter erhalten, dessen Knoten den Zentren der Partikel entsprechen, aus denen der Kristall besteht.
Wenn wir uns das an jeder Stelle vorstellen Bei 0 Co usw. wird das gleiche Gitter wiederhergestellt wie in A 0 , als Ergebnis dieser Konstruktion wird ein räumliches Gitter erhalten, das in gewissem Sinne die geometrische Struktur des Kristalls ausdrückt.
Was sind kristalle
Die vom großen russischen Kristallographen E. S. Fedorov entwickelte Theorie der räumlichen Gitter erhielt eine brillante Bestätigung bei der Untersuchung der Struktur von Kristallen mit Röntgenstrahlen. Diese Studien liefern nicht nur Bilder von räumlichen Gittern, sondern auch die genauen Längen der Lücken zwischen den Teilchen, die sich in ihren Knoten befinden.

Reis. 19. Diamantstruktur
Es stellte sich heraus, dass es mehrere Arten von räumlichen Gittern gibt, die sich sowohl in der Art der Anordnung der Teilchen als auch in ihrer chemischen Natur unterscheiden.
Wir bemerken die folgenden Arten von räumlichen Gittern:
Atomare Strukturgitter. An den Knoten dieser Gitter befinden sich Atome beliebiger Substanzen oder Elemente, die in einem Kristallgitter direkt miteinander verbunden sind. Dieser Gittertyp ist typisch für Diamant, Zinkblende und einige andere Mineralien (siehe Abb. 19 und 20).
Ionenstrukturgitter. An den Knoten dieser Gitter befinden sich Ionen, also Atome, die eine positive oder negative Ladung haben.
Ionengitter sind bei anorganischen Verbindungen wie Alkalimetallhalogenen, Silikaten usw. üblich.
Ein hervorragendes Beispiel ist das Gitter von Steinsalz (NaCl) (Abb. 21). Darin wechseln sich Natriumionen (Na) in drei zueinander senkrechten Richtungen mit Chloridionen (Cl) in Abständen von 0,28 Millimikron ab.

Reis. 20. Struktur von Zinkblende
In kristallinen Substanzen mit ähnlicher Struktur sind die Lücken zwischen Atomen in einem Molekül gleich den Lücken zwischen Molekülen, und der eigentliche Begriff eines Moleküls verliert für solche Kristalle seine Bedeutung. Auf Abb. 20 hat jedes Natriumion
von oben, unten, rechts, links, davor und dahinter in gleichen Abständen davon je ein Chlorion, das sowohl zu diesem „Molekül“ als auch zu benachbarten „Molekülen“ gehört und mit dem man nichts sagen kann welches bestimmte Chlorion dieser sechs ein Molekül darstellt oder es beim Übergang in einen gasförmigen Zustand darstellen würde.
Neben den oben beschriebenen Arten gibt es molekulare Strukturgitter, in deren Knoten keine Atome oder Ionen, sondern separate, elektrisch neutrale Moleküle vorhanden sind. Molekülgitter sind besonders typisch für verschiedene organische Verbindungen oder beispielsweise für "Trockeneis" - kristallines CO 2.

Reis. 21. Kristallgitter aus Steinsalz
Schwache ("Rest") Bindungen zwischen den Struktureinheiten solcher Gitter bestimmen die geringe mechanische Festigkeit solcher Gitter, ihre niedrigen Schmelz- und Siedepunkte. Es gibt auch Kristalle, die verschiedene Arten von Gittern kombinieren. In einigen Richtungen sind die Bindungen von Partikeln ionisch (Valenz) und in anderen molekular (Rest). Diese Struktur führt zu unterschiedlicher mechanischer Festigkeit in verschiedenen Richtungen, was eine scharfe Anisotropie der mechanischen Eigenschaften verursacht. Daher spalten sich Molybdänit (MoS 2 )-Kristalle leicht entlang der Pinacoid- (0001)-Richtung und verleihen den Kristallen dieses Minerals ein schuppiges Aussehen, ähnlich wie Graphitkristalle, wo eine ähnliche Struktur gefunden wird. Der Grund für die geringe mechanische Festigkeit in Richtung senkrecht zu (0001) ist das Fehlen ionischer Bindungen in dieser Richtung. Die Integrität des Gitters wird hier nur durch Bindungen molekularer (Rest-) Natur aufrechterhalten.
Wenn Sie alle oben genannten Punkte berücksichtigen, ist dies einfach eine Parallele zwischen der inneren Struktur einer amorphen Substanz einerseits und einer kristallinen andererseits:
1. In einer amorphen Substanz sind die Teilchen ungeordnet angeordnet, als ob sie den teilweise chaotischen Zustand der Flüssigkeit fixieren würden; daher nennen einige Forscher beispielsweise unterkühlte Flüssigkeiten.
2. In einer kristallinen Substanz sind die Teilchen geordnet angeordnet und nehmen an den Knoten des Raumgitters eine bestimmte Position ein.
Der Unterschied zwischen kristalliner und glasiger (amorpher) Materie kann mit dem Unterschied zwischen einer disziplinierten Militäreinheit und einer zerstreuten Menge verglichen werden. Natürlich ist der kristalline Zustand stabiler als der amorphe Zustand, und eine amorphe Substanz wird sich leichter auflösen, chemisch reagieren oder schmelzen. Natürliche neigen immer dazu, eine kristalline Struktur anzunehmen, beispielsweise „kristallisieren“ (amorphe Kieselsäure) wird schließlich zu Chalcedon - kristalline Kieselsäure.
Eine Substanz in kristallinem Zustand nimmt gewöhnlich ein etwas kleineres Volumen ein als in amorpher Form und hat ein größeres spezifisches Gewicht; Beispielsweise nimmt die Albit-Feldspat-Zusammensetzung NaAlSi 3 O 8 in amorphem Zustand 10 Kubikmeter ein. Einheiten und im Kristall - nur 9; eines cm 3 kristallines Siliziumdioxid (Quarz) wiegt 2,54 G, und das gleiche Volumen an Quarzglas (Quarzglas) beträgt nur 2,22 G. Ein Sonderfall ist Eis, das ein geringeres spezifisches Gewicht hat als in gleicher Menge eingenommen.
UNTERSUCHUNG VON KRISTALLEN MIT RÖNTGEN STRAHLEN
Die Frage nach den Ursachen von Regelmäßigkeiten in der Verteilung physikalischer Eigenschaften in einer kristallinen Substanz, die Frage nach der inneren Struktur von Kristallen wurde erstmals 1749 von M. V. am Beispiel des Salpeters versucht. Diese Frage wurde dann bereits Ende des 18. Jahrhunderts weiter entwickelt. Der französische Kristallograph Ayui. Ayui schlug vor, dass jede Substanz eine spezifische kristalline Form hat. Diese Position wurde später durch die Entdeckung der Phänomene der Isomorphie und Polymorphie widerlegt. Diese Phänomene, die in der Mineralogie eine wichtige Rolle spielen, werden wir etwas später betrachten.
Dank der Arbeit des russischen Kristallographen E. S. Fedorov und einiger anderer Kristallographen wurde die Theorie der räumlichen Gitter, die im vorigen Kapitel kurz umrissen wurde, mathematisch entwickelt, und basierend auf dem Studium der Form von Kristallen wurden mögliche Arten von räumlichen Gittern abgeleitet ; aber erst im 20. Jahrhundert wurde diese Theorie dank der Untersuchung von Kristallen durch Röntgenstrahlen experimentell überprüft und brillant bestätigt. Einer Reihe von Physikern wie Laue, Braggum, G. V. Wulf und anderen ist es gelungen, mithilfe der Theorie der räumlichen Gitter mit absoluter Sicherheit nachzuweisen, dass sich in einigen Fällen Atome an den Knoten von Kristallgittern befinden, in anderen Fällen Moleküle oder Ionen.
Die 1895 von Röntgen entdeckten Strahlen, die seinen Namen tragen, stellen eine der Arten von Strahlungsenergie dar und sind in vielerlei HinsichtSie ähneln Lichtstrahlen und unterscheiden sich von ihnen nur in ihrer Wellenlänge, die mehrere tausend Mal kleiner ist als die Lichtwellenlänge.

Reis. 22. Schema zum Erhalten eines Röntgenbeugungsmusters eines Kristalls unter Verwendung der Laue-Methode:
A - Röntgenröhre; B - Zwerchfell; C - Kristall; D - fotografische Platte
1912 verwendete Laue einen Kristall, in dem die Atome in einem räumlichen Gitter angeordnet sind, als Beugungsgitter, um Röntgeninterferenz zu erhalten. In seiner Forschung wurde ein schmaler Strahl paralleler Röntgenstrahlen (Abb. 22) durch einen dünnen Kristall aus Zinkblende C geleitet. In einiger Entfernung vom Kristall und Senkrecht zum Strahlenbündel wurde eine fotografische Platte D aufgestellt, die durch Bleischirme vor der direkten Einwirkung seitlicher Röntgenstrahlen und vor Tageslicht geschützt war.
Bei längerer Exposition über mehrere Stunden erhielten die Experimentatoren ein ähnliches Bild wie in Abb. 23.
Für Lichtstrahlen, die im Vergleich zur Größe von Atomen eine große Wellenlänge haben, spielen die Atomgitter des Raumgitters die Rolle von praktisch durchgehenden Ebenen, und die Lichtstrahlen werden vollständig von der Oberfläche des Kristalls reflektiert. Viel kürzere Röntgenstrahlen, die von zahlreichen Atomgittern reflektiert werden, die sich in bestimmten Abständen voneinander befinden und in die gleiche Richtung gehen, werden sich gegenseitig stören, schwächen und dann verstärken. Auf einer fotografischen Platte, die in ihren Weg gestellt wird, ergeben die verstärkten Strahlen während einer Langzeitbelichtung schwarze Flecken, die regelmäßig angeordnet sind und in enger Verbindung mit der inneren Struktur des Kristalls stehen, dh mit seinem Atomnetzwerk und mit den Merkmalen der einzelnen lokalisierten Atome drin.
Nimmt man eine Platte, die in einer bestimmten kristallographischen Richtung aus einem Kristall geschnitten ist, und führt damit das gleiche Experiment durch, dann wird auf dem Röntgenbild ein Muster sichtbar, das der Symmetrie der Kristallstruktur entspricht.
Die dichteren Atomnetzwerke entsprechen den dunkelsten Flecken. Spärlich mit Atomen besetzte Flächen ergeben Schwachstellen oder fast keine. Der zentrale Fleck auf einem solchen Röntgenbild wird aus Röntgenstrahlen erhalten, die die Platte passiert haben

Reis. 23. Röntgenbeugung eines Steinsalzkristalls entlang der Achse 4. Ordnung
auf geradem Weg; die verbleibenden Flecken bilden Strahlen, die von Atomgittern reflektiert werden.
Auf Abb. 23 zeigt eine Röntgenaufnahme eines Steinsalzkristalls, aus dem etwa eine Platte geschnitten wurde 3 mm Dicke parallel zur Fläche des Würfels. In der Mitte ist ein großer Fleck sichtbar - eine Spur des zentralen Strahlenbündels.
Die Anordnung der kleinen Punkte ist symmetrisch und weist auf die Existenz einer Symmetrieachse 4. Ordnung und vier Symmetrieebenen hin.
Die zweite Abbildung (Abb. 24) zeigt ein Röntgenbeugungsmuster eines Calcitkristalls. Das Bild wurde in Richtung der Symmetrieachse 3. Ordnung aufgenommen. in Briefen Ö die Enden der Symmetrieachsen 2. Ordnung sind angedeutet.
Gegenwärtig werden verschiedene Methoden verwendet, um die Struktur kristalliner Körper zu untersuchen. Ein wesentliches Merkmal des oben kurz beschriebenen Laue-Verfahrens ist die Verwendung von nur großen Kristallen, die in Bezug auf den durchlaufenden Röntgenstrahl präzise orientiert sind.
Wenn große Kristalle nicht verwendet werden können, wird normalerweise die "Pulvermethode" (Debye-Scherer-Methode) verwendet. Der große Vorteil dieser Methode ist, dass keine großen Kristalle benötigt werden. Vor der Prüfung wird die Prüfsubstanz in der Regel in fein verteiltem Zustand in eine kleine Säule gepresst. Mit dieser Methode lassen sich nicht nur gepresste Pulver untersuchen, sondern auch fertige Metallproben in Form eines Drahtes bearbeiten, wenn deren Kristalle klein genug sind.
Bei Vorhandensein einer großen Anzahl von Kristallen kann eine Reflexion von jeder Fläche jedes Kristalls auftreten. Daher wird in dem durch die "Pulvermethode" erhaltenen Röntgenmuster normalerweise eine Reihe von Linien erhalten, die eine Eigenschaft der untersuchten Substanz angeben.
Durch die Untersuchung von Kristallen mit Röntgenstrahlen war es schließlich möglich, in den Bereich der tatsächlichen Lage von Molekülen, Ionen und Atomen im Inneren von Kristallen vorzudringen und nicht nur die Form des Atomgitters, sondern auch die Abstände zwischen ihnen zu bestimmen Teilchen, aus denen es besteht.
Die Untersuchung der Struktur von Kristallen mit Röntgenstrahlen ermöglichte es, die scheinbare Größe der Ionen zu bestimmen, aus denen dieser Kristall besteht. Das Verfahren zur Bestimmung des Werts des Radius eines Ions oder, wie sie gewöhnlich sagen, des Ionenradius, wird aus dem folgenden Beispiel deutlich. Die Untersuchung solcher Kristalle wie MgO, MgS und MgSe einerseits und MnO, MnS und MnSe andererseits ergab die folgenden interionischen Abstände:
Zum
MgO – 2,10 Å MnO – 2,24 Å
MgS – 2,60 Å und MnS – 2,59 Å
MgSe – 2,73 Å MnSa – 2,73 Å,
wobei Å den Wert von "Angström" bezeichnet, der einem Zehnmillionstel Millimeter entspricht.
Ein Vergleich der angegebenen Werte zeigt, dass für den interionischen Abstand in den MgO- und MnO-Verbindungen die Größen der Mg- und Mn-Ionen eine gewisse Rolle spielen. Bei anderen Verbindungen ist zu sehen, dass der Abstand zwischen den S- und Se-Ionen nicht von der Eingabe abhängtein weiteres Ion, das die Verbindungen verbindet, und die S- und Se-Ionen kommen miteinander in Kontakt, wodurch die dichteste Packung von Ionen entsteht.

Reis. 24. Röntgenbild eines Calcitkristalls auf der Achse 3. Ordnung
Die Berechnung ergibt für S -2 einen Ionenradius von 1,84 Å,
a für Se –2 – 1,93 Å. Wenn man die Ionenradien S -2 und Se -2 kennt, kann man auch die Ionenradien anderer Ionen berechnen. O 2 hat also einen ionischen
Radius gleich 1,32 Å. F -1 - 1,33 Å, Na + 1 -0,98 Å, Ca + 2 - 1,06,
K +1 - 1,33, Mg +2 -0,78 Å, Al +3 -0,57 Å, Si +4 - 0,39 Å usw. Der Wert des Ionenradius spielt eine große Rolle bei Isomorphismus und Polymorphismus, was in diskutiert wird die entsprechenden Rubriken.
Die Untersuchung der Röntgenstruktur von Mineralien hat die moderne Mineralogie erheblich vorangebracht, sowohl im Hinblick auf das Verständnis der Struktur von Mineralien als auch in Bezug auf die Beziehung ihrer Struktur und Zusammensetzung zu anderen wichtigen Eigenschaften wie Spaltung, Brechungsindex usw. Die Bedeutung der Die Untersuchung von Mineralien durch Röntgenstrahlen wird durch den folgenden Ausdruck schön ausgedrückt: Mineral insofern, als man ein Gebäude untersuchen kann, indem man es von außen betrachtet, und Chemiker versuchten, dieses Gebäude zu erkennen, indem sie es zerstörten und dann die Materialien, die dazu gehörten, separat untersuchten davon erlaubte uns die Röntgenbeugungsanalyse zum ersten Mal, das Gebäude zu betreten und seine innere Lage und Dekoration zu beobachten."
Artikel zum Thema Aufbau von Kristallen
KRISTALLSTRUKTUR, die Anordnung der Atome kristallin. in-va im Raum. max. die charakteristische Eigenschaft der Kristallstruktur ist eine dreidimensionale Periodizität (siehe Kristallzustand). Üblicherweise implizieren sie, wenn man von einer Kristallstruktur spricht, die zeitlich gemittelte Anordnung von Atomkernen (das sogenannte statische Modell); vollständigere Informationen umfassen Informationen über die Amplituden und Frequenzen von Atomschwingungen (dynamisches Modell) sowie über die Verteilung der Elektronendichte im Kernraum. Das Studium von Kristallstrukturen und deren Zusammenhang mit St. du in-in ist Gegenstand der Kristallchemie. Geom. Eigenschaften der Kristallstruktur, Daten zur Verteilung der Elektronendichte, Amplituden von Atomschwingungen (genauer gesagt quadratische Mittelverschiebungen aus Gleichgewichtspositionen) werden mit Methoden der Beugungsforschung (Röntgenbeugungsanalyse, Neutronenbeugung und Elektronenbeugung) ermittelt von Kristallen), Schwingungsfrequenzen - durch spektroskopische Methoden (IR, kombinatorische Streuung, inelastische Neutronenstreuung). Modellierung der Kristallstruktur. Die ideale Kristallstruktur ist durch unendliche Räume gekennzeichnet. Gitter, d.h. besteht aus identischen Elementarzellen. Letztere haben die Form von Parallelepipeden mit Seiten a, b, c und Winkeln a, b, g (Gitterparameter) und stehen in Kontakt mit ganzen Flächen. Bei echten Kristallen wird die Kristallstruktur immer durch Defekte sowie durch das Vorhandensein von Kristalloberflächen verzerrt. Manchmal wird anstelle des Begriffs "Kristallstruktur" der Begriff "Kristallgitter" verwendet; es ist jedoch vorzuziehen, letzteren einen anderen Inhalt zu geben (siehe Kristalle). Zur Beschreibung der Statik Modell der Kristallstruktur, es ist notwendig, seine Symmetrie anzugeben, die durch eine der räumlichen (Fedorov-) Gruppen, Gitterparameter und Koordinaten von Atomkernen in der Zelle ausgedrückt wird; diese Daten ermöglichen die Berechnung von Atomabständen und Bindungswinkeln. Die primäre Interpretation eines solchen Modells in Gegenwart von kovalenten Bindungen zwischen Atomen ist, dass die Atome in Übereinstimmung mit der Klassik durch Valenzstriche verbunden sind. die Theorie der Chem. Gebäude. Interatomare Abstände zeigen die richtige Art, Valenzstriche zu zeichnen: Normalerweise ist der Abstand A - B, der einer kovalenten Bindung entspricht, deutlich kürzer als der kürzeste Abstand zwischen den valenzfreien Atomen A und B. Wenn keine kovalenten Bindungen (ionisch, metallisch oder van der Waals vorherrschende interatomare Wechselwirkungen), stellt sich das Modell der Kristallstruktur in Form einer dichten Packung aus Kugeln gleicher Größe (einfach in-va) oder Kugeln aus mehreren dar.Reis. 1. Ellipsoide thermischer Schwingungen von Atomen in der Struktur m -acetylen-bis(cyclopentadiennickel) bei 300 K (a) und 77 K (6). In der Mitte befindet sich ein Acetylenmolekül, an den Seiten befinden sich Cyclopentadienmoleküle.
Sorten (z. B. Anionen bilden ein Paket, Kationen befinden sich in dessen Hohlräumen). Die Berücksichtigung der dreidimensionalen Verteilung der Elektronendichte p im Raum der kartesischen Koordinaten x, y, z führt zu einem Modell der Kristallstruktur, wonach die Atomkerne in eine kontinuierlich mit a verteilte elektronische Ladung „eingetaucht“ sind Dichte p. Modern Mit der präzisen Röntgenbeugungsanalyse können Sie die Merkmale der Funktion experimentell untersuchen r (x, y, z) und bestimmen die Änderung der Elektronendichte von Atomen im Kristall im Vergleich zur Elektronendichte r 0 Valenz ungebundene Atome, resultierend aus der Quantenchemie. Berechnungen. Diese Daten können sein. nützlich zum Festlegen von Bereichen der Lokalisierung von Valenz- und ungeteilten Elektronenpaaren, zum Nachweis von Ladungstransfer und anderen strukturellen Merkmalen von in-in mit kovalenten Bindungen sowie in-in, in denen gerichtete interatomare Wechselwirkungen auftreten. fehlen. Um die Dynamik der Atome in der Kristallstruktur harmonisch wiederzugeben. Annäherung, Atome werden als "thermische Ellipsoide" dargestellt, To-Roggen haben eine Spur. körperlich Bedeutung: mit einem Fix. mit einer Wahrscheinlichkeit p befindet sich der Atomkern zu jedem Zeitpunkt innerhalb oder auf der Oberfläche eines solchen Ellipsoids (Abb. 1). Richtung max. die Verlängerung des Ellipsoids entspricht der Richtung, in der das Atom die maximale Amplitudenschwingung ausführt, die Richtung des Maximums. Kompression entspricht den minimalen Amplitudenschwingungen. Normalerweise normieren sie auf die Wahrscheinlichkeit p = 1/2. Für ein gegebenes p hängen die Abmessungen der Ellipsoide von t-ry ab. Um die Form und Orientierung von atomaren thermischen Ellipsoiden quantitativ zu charakterisieren, geben Sie für jedes Atom 6 unabhängige Komponenten des symmetrischen Tensors des 2. Ranges an, deren Werte aus den Daten von Röntgenbeugungsstudien bestimmt werden. Die beschriebene Dynamik das Modell gibt keine Auskunft über den momentanen Aufbau des Kristalls und über die Sukzession. Änderung von Instant-Strukturen. Informationen dieser Art können aus inelastischen Neutronenstreuspektren gewonnen werden. Klassifizierung von Kristallstrukturen. Im Prinzip jeder kristallin. in-woo hat eine eigene Struktur. Allerdings haben unterschiedliche Substanzen oft bis zur Ähnlichkeit gleiche Kristallstrukturen (die sogenannte Isostruktur). Manchmal sind solche Substanzen in der Lage, Mischkristalle zu bilden (siehe Isomorphie). Andererseits ist die gleiche chem. Anschluss in verschiedenen Thermodynamiken Bedingungen und unter verschiedenen Herstellungsmethoden können unterschiedliche Kristallstrukturen haben (siehe Polymorphismus). Kristallstrukturen sind sehr vielfältig – von einfach (z. B. Diamant) bis zu extrem komplex (z. B. Bor). Mehrere Kristallstrukturen wurden untersucht. Zehntausende von In-In, einschließlich Proteinen und anderer komplexer Natur. Anschluss Für mehrere Hunderte von kristallinen in-in (sowohl anorg. als auch org.) wurde die Verteilung der Elektronendichte in Kristallen untersucht. Zu kristalline Strukturen unterscheiden nagomodesmisch (Koordination) und heterodesmisch. Im ersten Fall sind alle Atome durch dieselbe Chemikalie verbunden. Verbindungen, die Räume bilden. Gerüst (z. B. Diamant, Alkalimetallhalogenide). Letztere sind durch das Vorhandensein von Strukturfragmenten gekennzeichnet, in denen die Atome am stärksten verbunden sind. starke (meistens kovalente) Bindungen; Atome gehören zu dez. Fragmente sind viel schwächer verbunden. Fragmente können endliche Atomgruppen ("Inseln"), Ketten, Schichten, Gerüste sein; bzw. Insel-, Ketten-, Schicht- und Rahmenkristallstrukturen zuordnen. Fast alle org. haben Inselkristallstrukturen. comp. sowie Halogene, O 2, S, (NH 4) 2 SO 4 usw. Die Rolle der Inseln spielen Moleküle (siehe Molekülkristalle) oder mehratomige Ionen. Eine Kettenkristallstruktur hat beispielsweise eine der Modifikationen von Se, bei der die Atome in endlosen Spiralen verbunden sind. Graphit, BN, MoS 2 usw. haben eine Schichtstruktur Ein Beispiel für eine Rahmenkristallstruktur sind CaTiO 3 -Kristalle: Durch kovalente Bindungen verbundene Ti- und O-Atome bilden ein durchbrochenes Gerüst, in dessen Hohlräumen sich Ca-Atome befinden. Es sind Kristallstrukturen bekannt, in denen Strukturfragmente unterschiedlicher Art koexistieren. Somit sind die Kristalle des Komplexes Comm. N(CH 3) 4 bestehen aus "Inseln" - N(CH 3) 4 -Ionen und -Ketten, die aus Mn-Atomen gebildet werden, die durch verbrückende Cl-Atome verbunden sind. Oft gibt es Kristallstrukturen mit unvollständiger Ordnung, in denen einzelne Atome oder Strukturfragmente statistisch mehrere besetzen. mögliche Positionen (z. B. statistische Schichtung in CdI 2). In bestimmten Kristallstrukturen befinden sich bei ausreichend hoher Temperatur einzelne Atomgruppen oder sogar ganze Moleküle in einem nahezu freien oder verzögerten Rotationszustand. Je nach Art der Bindung zwischen Atomen oder Strukturbruchstücken unterscheidet man kovalente Kristalle, Ionenkristalle, Metallkristalle und Van-der-Waals-Kristalle. Zur letzten Gruppe gehören insbesondere Molekülkristalle. Diese Einteilung (wie auch die Einteilung chemischer Bindungen in Typen) ist jedoch bedingt, typische Vertreter verschiedener Gruppen unterscheiden sich beispielsweise stark in St. you. entsprechend der Energie der Struktur (die zum Trennen erforderliche Energie).
MUTIGES GITTER
Konstruktionsschema
BRAVE LATTICES, 14 dreidimensionale geometrische Gitter, die alle möglichen Typen der Translationssymmetrie von Kristallen charakterisieren. Brave-Gitter werden durch die Wirkung der Übertragungs-(Translations-)Operation an irgendeinem Punkt des Kristalls gebildet.
O. Brave zeigte 1848, dass die ganze Vielfalt der Kristallstrukturen durch 14 Arten von Gittern beschrieben werden kann, die sich in Form von Elementarzellen und Symmetrie unterscheiden und in 7 kristallographische Syngonien unterteilt sind. Diese Gitter wurden Bravais-Gitter genannt.
Bravais-Gitter unterscheiden sich in der Symmetrie der Elementarzelle, also im Verhältnis ihrer Kanten und Ecken, sowie in ihrer Zentrierung.
Drei Bedingungen werden verwendet, um eine Bravais-Zelle auszuwählen:
Die Symmetrie der Einheitszelle muss der Symmetrie des Kristalls entsprechen, genauer gesagt der höchsten Symmetrie der Syngonie, zu der der Kristall gehört. Die Elementarzellenränder müssen Translationen des Gitters sein;
Die Elementarzelle muss die maximal mögliche Anzahl rechter Winkel oder gleicher Winkel und gleicher Kanten enthalten;
Die Elementarzelle muss ein Mindestvolumen haben.
Je nach Art der gegenseitigen Anordnung der Hauptübersetzungen oder der Anordnung der Knoten werden alle Kristallgitter in vier Typen unterteilt: primitiv ( R), basiszentriert ( AUS), körperzentriert ( ich), gesichtszentriert ( F).
Im Primitiven R-Zellgitterknoten befinden sich nur an den Eckpunkten der Zelle, in einem Körper zentriert ich-cell – ein Knoten in der Mitte der Zelle, flächenzentriert F-cell – ein Knoten in der Mitte jeder Fläche, in Basis-zentriert AUS-cell - ein Knoten in der Mitte eines Paars paralleler Flächen.
Der Satz von Koordinaten der in der Elementarzelle enthaltenen Knoten wird als Basis der Zelle bezeichnet. Die gesamte Kristallstruktur kann erhalten werden, indem die Basisknoten durch einen Satz von Translationen der Bravais-Zelle wiederholt werden.
Bei einigen Syngonien kann eine Elementarzelle Knoten nicht nur in den Ecken, sondern auch in der Mitte der Zelle, allen oder einigen der Flächen enthalten. Dabei ist eine translatorische Übertragung nicht nur auf die Perioden der Elementarzelle, sondern auch auf die Hälfte der Diagonalen der Zellflächen bzw. Raumdiagonalen möglich. Zusätzlich zur obligatorischen Translationsinvarianz kann sich das Gitter unter anderen Transformationen, zu denen Rotationen, Reflexionen und Inversionen gehören, in sich selbst verwandeln. Es sind diese zusätzlichen Symmetrien, die den Typ des Bravais-Gitters bestimmen und ihn von anderen unterscheiden.
Mutige Gittertypen:
Kubisch: primitiv, körperzentriert und gesichtszentriert;
Sechseckig, dreieckig;
Tetragonal: primitiv und volumenzentriert;
Rhombisch: primitiv, basis-, volumen- und flächenzentriert;
Monoklin: primitiv und basenzentriert;
Triklinik.

Syngonie(aus dem Griechischen σύν, „zusammen, nebeneinander“, und γωνία, „Winkel“ - wörtlich „ähnlicher Winkel“) - Klassifizierung von kristallographischen Symmetriegruppen, Kristallen und Kristallgittern in Abhängigkeit vom Koordinatensystem (Koordinatenbezugssystem ). Symmetriegruppen mit einem einzigen Koordinatensystem werden zu einer Syngonie zusammengefasst.
Kristalle, die zu derselben Syngonie gehören, haben ähnliche Ecken und Kanten von Elementarzellen.
Triklinik: (\displaystyle a\neq b\neq c), (\displaystyle \alpha \neq \beta \neq \gamma \neq 90^(\circ ))
Monoklin: (\displaystyle a\neq b\neq c), (\displaystyle \alpha =\gamma =90^(\circ ),\beta \neq 90^(\circ ))
Rhombisch: (\displaystyle a\neq b\neq c), (\displaystyle \alpha =\beta =\gamma =90^(\circ ))
Tetragonal: (\displaystyle a=b\neq c), (\displaystyle \alpha =\beta =\gamma =90^(\circ ))
Hexagonal: (\displaystyle a=b\neq c), (\displaystyle \alpha =\beta =90^(\circ ),\gamma =120^(\circ ))
Kubisch: (\displaystyle a=b=c), (\displaystyle \alpha =\beta =\gamma =90^(\circ ))

Hauptmerkmale von Kristallstrukturen
Kristalline Materialien sind durch das Vorhandensein einer Fernordnung gekennzeichnet, was charakteristisch ist. dadurch, dass darin ein bestimmtes Volumen unterschieden werden kann, wiederholt sich die Anordnung des Atoms in dem gesamten Volumen.
In amorphen Matten gibt es eine Nahordnung, Kat. Charakter Themen. dass es keine Wiederholung von Bänden gibt.
Kris. die Struktur lässt sich bequem mit Hilfe von Z beschreiben X ein dimensionales Gitter aus geraden Limetten, die den Raum in gleich große Parallelepipede unterteilen. Das Kreuzen von Linien ist ein Bild von dreidimensionalen Räumen. Gitter. Gitterknoten entsprechen in der Regel der Anordnung von Atomen in einem Kristall. Das Atom schwingt
um diese Positionen. Wenn es in einem solchen räumlichen Gitter möglich ist, ein bestimmtes Volumen herauszuheben, indem man es in 3 Richtungen bewegt. ermöglicht es Ihnen, den gesamten Kristall auszurichten, dann gov. Dass ein Element, eine Zelle gefunden wurde.
Das Zellelement ist normalerweise durch 6 Parameter gekennzeichnet: a, b, c - die Länge der Kanten des Parallelepipeds, α, β, γ.
Die Form des Zellelements bestimmt das kristallographische Koordinatensystem - Syngonie. Als Achsen werden die Richtungen der Kanten – Elemente, Zellen gewählt, und die Kanten selbst sind die Maßeinheiten. Die Anzahl der rechten Winkel und gleichen Seiten muss maximal sein, und das Volumen der Zellelemente muss minimal sein.