Регулярная трисомия по 18 хромосоме. Диагноз Синдром Эдвардса – что это такое
Читайте также
Большинство женщин хорошо знают, что такое синдром Дауна. И во время беременности многие узнают, что очень редко, но встречается и другое хромосомное нарушение, называемое Синдром Эдвардса. И многие обеспокоены, как узнать насколько высок риск рождения ребенка с синдромом Эдвардса и как диагностируется такая патология при беременности?
Что такое Синдром Эдвардса?
Синдром Эдвардса - это генетическое заболевание, характеризующееся дублированием (трисомией) ХVIII хромосомы и проявляющееся целым рядом характерных пороков развития у плода во время беременности, часто приводящих к смерти ребенка или его инвалидизации. То есть у ребенка вместо 46 хромосом образуется 47, эта лишняя хромосома дает другое название болезни - трисомия 18. Синдром был назван в честь исследователя Джона Эдвардса, который впервые описал его в 1960 году.
Почему возникает синдром Эдвардса - причины патологии
Даже если родители здоровы, и в семейном анамнезе нет такой патологии, ребенок с 18 хромосомой может родиться у любой женщины.
Как известно, в каждой человеческой клетке находится 46 хромосом, а в женских и мужских половых клетках по 23 хромосомы, которые, соединяясь при оплодотворении яйцеклетки, также дают в сумме 46 хромосом. Если говорить о синдроме Эдвардса, причины его появления неизвестны.
В настоящее время известно только то, что в результате определенных генетических мутаций появляется лишняя 47–я хромосома (дополнительная хромосома в 18–й паре хромосом, которых таким образом становится не 2, а 3).
В 95% всех случаев развития синдрома Эдвардса в клетках находится именно лишняя 18–я хромосома (трисомия), однако в 2% наблюдается только «удлинение» 18–й хромосомы (транслокация), когда общее число хромосом остается нормальным и равняется 46.
В 3% случаев синдрома Эдвардса имеет место «мозаичная трисомия», когда дополнительная 47–я хромосома обнаруживается в организме не во всех клетках, а лишь определенной его части. Клинически все 3 варианта синдрома Эдвардса протекают практически одинаково, однако первый вариант может отличаться более тяжелым течением заболевания.
Как часто встречается эта патология?
Дети с синдромом Эдвардса погибают на этапе внутриутробного развития приблизительно в 60% случаев. Несмотря на это, среди генетических заболеваний данный синдром у выживших младенцев является достаточно распространенным, по частоте встречаемости уступая только синдрому Дауна. Распространенность синдрома Эдвардса составляет 1 случай на 3 - 8 тысяч детей.
Считается, что у девочек данное заболевание встречается в 3 раза чаще, а риск синдрома Эдвардса значительно возрастает, если беременной женщине 30 и более лет.
Смертность при синдроме Эдвардса на первом году жизни составляет около 90%, причем средняя продолжительность жизни при тяжелом течении заболевания у мальчиков - 2–3 месяца, а девочек - 10 месяцев, а до взрослого состояния доживают лишь единицы. Чаще всего дети с синдромом Эдвардса умирают от удушья, пневмонии, сердечно–сосудистой недостаточности или кишечной непроходимости - осложнений, вызванных врожденными пороками развития.
Как проявляется синдром у ребенка?
Признаки синдрома Эдвардса можно разделить на несколько больших групп:
К первой группе относятся симптомы, характерные для внешнего вида ребенка:
- При рождении низкая масса тела (около 2 100 – 2200 граммов)
- Непропорционально маленькая голова
- Дефекты развития верхней или нижней челюстей (микрогнатия)
- Искажение формы лица и формирование неправильного прикуса
- «Волчья пасть» (расщелина твердого неба) или «заячья губа» (расщелина верхней губы)
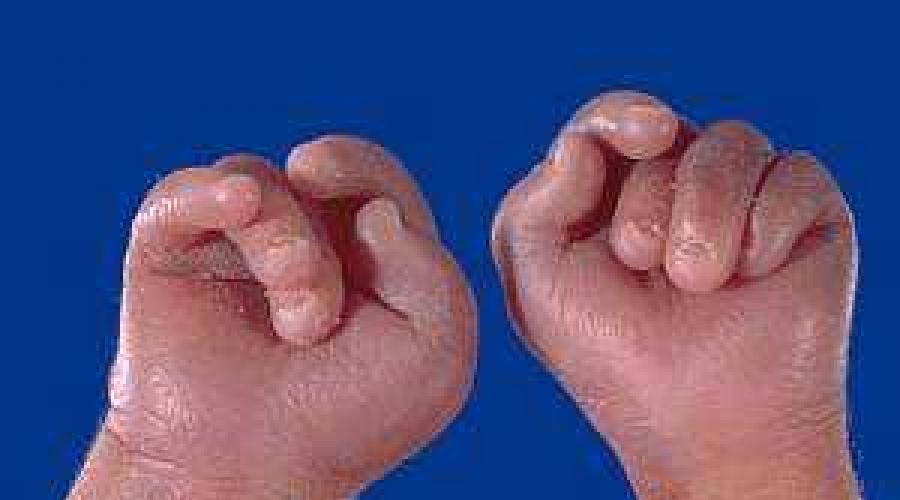
- Пальцы кисти сжаты, в кулаке располагаются неровно
- Низкая посадка ушей
- Перепончатость или полное слияние пальцев нижних конечностей
- Врожденная косолапость
- «Стопа–качалка»
- Относительно малых размеров ротовая щель (микростомия)
Ко второй группе можно отнести признаки нарушения работы внутренних органов, моторики и нервно–психического развития:
- Наличие врожденных пороков сердца, например, открытого овального отверстия, дефекта межжелудочковой перегородки, открытого артериального протока и т.п.
- Развитие паховых или пупочных грыж.
- Органы пищеварения: гастроэозофагиальная рефлюксная болезнь, нарушение глотательного и сосательного рефлекса, атрезия пищевода или заднего прохода, меккелев дивертикул, нарушение расположения кишечника.
- Центральная нервная система: задержка нервно–психического развития, умственная отсталость, недоразвитость мозжечка, мозолистого тела, сглаживание или атрофия мозговых извилин.
- Мочеполовая система: крипторхизм, гипоспадия у мальчиков, гипертрофия клитора, недоразвитость яичников у девочек, независимо от пола - подковообразная или сегментированная почка, удвоение мочеточников.
- Косоглазие, сколиоз, атрофия мышц.
Как узнать патологию во время беременности — диагностика
Синдром Патау, Эдвардса и другие трисомии лучше всего выявить до рождения малыша. Как правило, пренатальная диагностика данного синдрома проводится в 2 этапа:
- На сроке в 11–13 недель (скрининг, в основе которого - проведение различных биохимических анализов у женщины).
- Определение кариотипа плода у беременных их группы риска.
В 11–13 недель в крови женщины определяется уровень некоторых белков крови: β–ХГЧ (β–субъединица хорионического гормона человека) и плазменного протеина А ассоциированного с беременностью. Затем с учетом этих данных, возраста беременной женщины рассчитывается риск рождения ребенка с синдромом Эдвардса, и формируется группа риска беременных.
Далее в группе риска на более позднем сроке берется материал у плода для постановки точного диагноза: в 8–12 недель это биопсия ворсин хориона, в 14–18 - амниоцентез (изучение околоплодных вод), спустя 20 недель - кордоцентез (внутриутробное взятие крови из пуповины плода с УЗИ–контролем). После этого в полученном материале определяют наличие или отсутствие дополнительной 18–й хромосомы с помощью КФ–ПЦР (количественной флуоресцентной полимеразной цепной реакции).
Если беременная не прошла генетическое скрининг–обследование, то на более поздних сроках предварительная диагностика синдрома Эдвардса осуществляется с помощью УЗИ. Прочие косвенные признаки, на основании которых можно заподозрить синдром Эдвардса на более поздних сроках:
- Наличие аномалий развития костей и мягких тканей головы («волчья пасть», микроцефалия, низкая посадка ушных раковин, «заячья губа» и т.п.).
- Обнаружение пороков со стороны сердечно–сосудистой, мочеполовой системы, а также опорно–двигательного аппарата.
Диагностические признаки синдрома у ребенка
После рождения ребенка опорными диагностическими признаками наличия синдрома Эдвардса являются следующие:

Признаки характерной дерматографической картины:
- неразвитая на пальцах дистальная сгибательная складка
- наличие в 1/3 случаев поперечной ладонной борозды
- дуги на подушечках пальцев рук
- изменение кожного рисунка ладони: дистальное расположение осевого трирадиуса и увеличение гребневого счета.
Синдром Эдвардса на УЗИ — кисты сосудистых сплетений
На ранних сроках синдром Эдвардса на УЗИ заподозрить крайне трудно, однако в 12 недель беременности уже выявляют характерные для него симптомы косвенного характера:
- Признаки задержки развития плода
- Брадикардия (снижение у плода частоты сердечных сокращений)
- Омфалоцеле (наличие грыжи брюшной полости)
- Отсутствие визуализации косточек носа
- В пуповине одна, а не 2 артерии
Также на УЗИ могут быть обнаружены кисты сосудистых сплетений, представляющих собой полости с содержащейся в них жидкостью. Сами по себе они не несут угрозу для здоровья и исчезают к 26–недельному сроку беременности. Однако такие кисты очень часто сопровождают различные генетические заболевания, например, синдром Эдвардса (в данном случае кисты обнаруживаются у 1/3 детей, страдающих этой патологией), поэтому при обнаружении таких кист врач направит беременную женщину на обследование в генетическую консультацию.
Так как дети с синдромом Эдвардса редко доживают до года, то сначала лечение направлено на коррекцию тех пороков развития, которые опасны для жизни:
- восстановление прохода пищи при атрезии кишечника или анального отверстия
- кормление через зонд в случае отсутствия глотательного и сосательного рефлексов
- антибактериальная и противовоспалительная терапия при воспалении легких
При относительно благоприятном течении заболевания происходит коррекция некоторых аномалий и пороков развития: хирургическое лечение «волчьей пасти», пороков сердца, паховой или пупочной грыжи, а также симптоматическое медикаментозное лечение (назначение слабительных при запорах, «пеногасителей» при метеоризме и т.п.). 
Дети с синдромом Эдвардса склонны к таким заболеваниям, как:
- средний отит
- рак почки (опухоль Вильмса)
- пневмония
- конъюнктивит
- легочная гипертензия
- апноэ
- повышенное артериальное давление
- фронтиты, синуситы
- инфекции мочеполовой системы
Поэтому лечение больных с синдромом Эдвардса включает своевременную диагностику и терапию данных заболеваний.
Прогноз для ребенка
В большинстве случаев прогноз неблагоприятный. Единицы детей с синдромом Эдвардса, которые доживают до взрослого возраста, имеют серьезные умственные отклонения и постоянно требуют постороннего ухода и контроля. Однако при соответствующих занятиях они способны реагировать на утешение, улыбаться, самостоятельно принимать пищу, а также взаимодействовать с воспитателями, приобретая различные умения и навыки.
Синдром Эдвардса

Синдром Эдвардса — хромосомное заболевание, обусловленное трисомией по 18-ой хромосоме и сопровождающееся множественными пороками развития. Для синдрома Эдвардса характерны своеобразные фенотипические признаки (долихоцефалическая форма черепа, микрофтальмия, недоразвитие ушных раковин, микроретрогнатия и др.), аномалии опорно-двигательной, сердечно-сосудистой, пищеварительной, мочеполовой системы, ЦНС. Синдром Эдвардса может быть диагностирован на этапе беременности (УЗИ-скрининг, инвазивная пренатальная диагностика) либо уже после рождения ребенка на основании внешних признаков и цитогенетического исследования. Дети с синдромом Эдвардса нуждаются в симптоматическом лечении и хорошем уходе.
Синдром Эдвардса

Синдром Эдвардса – количественная хромосомная аберрация, при которой имеет место частичная или полная трисомия по 18 аутосоме. Синдром получил название по имени генетика J. Edwards, подробно описавшего заболевание в 1960 г. и выделившего свыше 130 характерных для данной патологии симптоматических дефектов. Синдром Эдвардса – второе по распространенности хромосомное заболевание после синдрома Дауна; частота рождения детей с синдромом Эдвардса составляет 1:5000-7000. Примерно три четверти всех больных синдромом Эдвардса – девочки; предполагается, что большая часть беременностей плодом мужского пола заканчивается внутриутробной гибелью и самопроизвольным абортом.
Причины синдрома Эдвардса
Развитие синдрома Эдвардса объясняется хромосомными нарушениями, происходящими на стадии гаметогенеза (овогенеза или сперматогенеза) либо дробления зиготы и приводящими к увеличению числа хромосом 18-й пары. В 80-90% случаев цитогенетические варианты синдрома Эдвардса представлены простой трисомией 18, реже — мозаичной формой или несбалансированными перестройками (транслокациями).
Причиной полной трисомии служит мейотическое нерасхождение хромосом. Практически во всех случаях лишняя хромосома является материнской по происхождению. Этот вариант синдрома Эдвардса является наиболее тяжелым по своим проявлениям и неблагоприятным в плане прогноза. Возникновение мозаицизма связано с нерасхождением хромосом на ранней стадии дробления зиготы. В этом случае лишнюю хромосому будут содержать не все клетки плода, а лишь их часть. Транслокация – присоединение части 18-ой хромосомы к другой паре может произойти как в процессе созревания гамет, так и после оплодотворения. При этом клетки организма содержат две гомологичные 18-е хромосомы и ее дополнительную часть, прикрепленную к другой хромосоме.
Как и в случае с синдромом Дауна, возраст матери является наиболее значимым риск-фактором рождения ребенка с синдромом Эдвардса. В редких случаях у родителей может выявляться носительство сбалансированной транслокации.
Симптомы синдрома Эдвардса
Во время беременности наблюдается многоводие, слабая активность плода, маленькая плацента, единственная пупочная артерия. Ребенок с синдромом Эдвардса рождается с низкой массой тела (около 2170 г) и пренатальной гипотрофией при доношенной или даже переношенной беременности. У части детей определяется состояние асфиксии при рождении.
У новорожденных с синдромом Эдвардса имеются характерные фенотипические признаки, позволяющие предположить данную хромосомную патологию. В первую очередь обращает на себя внимание долихоцефалическая форма черепа с преобладанием продольного размера над поперечным, низкий лоб, выступающий затылок, микрогнатия, маленький рот, микрофтальмия. У детей с синдромом Эдвардса часто встречаются расщелины верхней губы и нёба, эпикант, птоз, экзофтальм, косоглазие, короткая шея с избыточной кожной складкой. Типичные деформации ушных раковин включают маленькие мочки, отсутствие козелков, узкие слуховые проходы, низкое расположение ушей.
Внешний облик детей дополняется характерными для синдрома Эдвардса деформациями скелета — скрещенными пальцами кистей, укороченной грудиной, аномалиями ребер, врожденным вывихом бедра, косолапостью, «стопой-качалкой», синдактилией стоп и пр. У многих детей имеются гемангиомы и папилломы кожи.
При синдроме Эдвардса имеются множественные тяжелые аномалии со стороны практически всех систем организма. Врожденные пороки сердца могут быть представлены дефектами межжелудочковой и межпредсердной перегородок, коарктацией аорты, транспозицией магистральных сосудов, дисплазией клапанов, тетрадой Фалло, аномальным дренажом легочных вен, декстракардией и др. При синдроме Эдвардса может выявляться патология развития желудочно-кишечного тракта: диафрагмальные, пупочные и паховые грыжи, дивертикул Меккеля, трахеопищеводные свищи, пилоростеноз, атрезия подвздошной кишки и ануса. Наиболее частыми аномалиями мочеполовой системы у детей с синдромом Эдвардса служат подковообразная почка, гидронефроз, дивертикулы мочевого пузыря, гипоспадия и крипторхизм (у мальчиков), двурогая матка, внутриматочная перегородка и гипертрофия клитора (у девочек).
Пороки развития центральной нервной системы характеризуются наличием микроцефалии, менингомиелоцеле, гидроцефалии, аномалии Арнольда-Киари, кист арахноидального сплетения, гипоплазии мозжечка и мозолистого тела. У всех выживших детей с синдромом Эдвардса имеются интеллектуальные нарушения — олигофрения в степени глубокой имбецильности или идиотии.
Новорожденные с синдромом Эдвардса испытывают трудности с сосанием, глотанием и дыханием, из-за чего им требуется зондовое питание или длительная ИВЛ. Дети с синдромом Эдвардса, как правило, погибают на первом году жизни из-за тяжелых врожденных пороков развития и связанных с ними осложнений (сердечно-сосудистой и дыхательной недостаточности, пневмонии, кишечной непроходимости и т. д.).
Диагностика синдрома Эдвардса
Важнейшей задачей диагностики служит антенатальное выявление синдрома Эдвардса у плода, поскольку данная патология является медицинским показанием для искусственного прерывания беременности. Заподозрить наличие синдрома Эдвардса можно в процессе УЗИ плода и допплерографии маточно-плацентарного кровотока по косвенным признакам (множественным аномалиям развития плода, агенезии пупочной артерии, малой величине плаценты, многоводию и пр.).
Наибольшую диагностическую значимость имеет стандартный пренатальный скрининг, включающий анализ крови на сывороточные маркеры: βХГЧ и PAPP на 11-13 неделе беременности; βХГЧ, альфа-фетопротеин и свободный эстриола на 20-24 неделе гестации.
При оценке степени риска рождения ребенка с синдромом Эдвардса учитываются данные биохимического и ультразвукового скрининга, срок беременности, возраст и масса тела женщины. Беременным, попадающим в группу высокого риска, предлагается проведение инвазивной дородовой диагностики (биопсии хориона, амниоцентеза, кордоцентеза) с последующим кариотипированием плода.
В случае рождения живого ребенка с синдромом Эдвардса необходимо как можно более раннее всестороннее обследование, направленное на выявление тяжелых пороков развития. Новорожденный с синдромом Эдвардса должен быть осмотрен неонатологом, детским кардиологом, детским неврологом, детским хирургом, детским ортопедом, детским урологом и др. Наиболее важными диагностическими исследованиями, которые должны быть выполнены ребенку с синдромом Эдвардса в первые часы жизни, служат эхокардиография, УЗИ органов брюшной полости и УЗИ почек.
Лечение синдрома Эдвардса
Поскольку в большинстве случаев аномалии развития оказываются несовместимыми с жизнью, лечение детей с синдромом Эдвардса сводится к оказанию симптоматической помощи, направленной на поддержание физиологических функций, продление жизни и улучшение ее качества. Хирургическая коррекция врожденных пороков, как правило, является рискованной и неоправданной.
Поскольку дети с синдромом Эдвардса ослаблены и подвержены частой заболеваемости инфекциями мочевыводящих путей, средним отитом, конъюнктивитом, синуситами, пневмониями и пр., они нуждаются в тщательно организованном уходе, полноценном питании, регулярном наблюдении со стороны педиатра.
Прогноз и профилактика синдрома Эдвардса
Во всех случаях прогноз при синдроме Эдвардса крайне неблагоприятный: в среднем мальчики живут 2-3 месяца, девочки – 10 месяцев. До 1 года доживает лишь 10% больных, до 10 лет – не более 1%. Относительно благоприятные шансы в отношении выживания имеют дети с мозаичной формой синдрома Эдвардса.
Риск рождения ребенка с синдром Эдвардса теоретически существует в любой супружеской паре; известно, что такая вероятность выше у возрастных родителей (для женщин старше 45 лет – 0,7%). С целью своевременного выявления хромосомной патологии у плода не следует пренебрегать антенатальным скринингом, входящим в программу введения беременности.
Эдвардса синдром
Наша команда профессионалов ответит на ваши вопросы
Синдром Эдвардса (трисомия по хромосоме 18 ) — второе по частоте после синдрома Дауна хромосомное нарушение. Частота синдрома Эдвардса составляет 1:5000-1:7000 новорождённых. Девочки с синдромом Эдвардса рождаются в три раза чаще мальчиков.
«Золотым стандартом» выявления хромосомных нарушений во всем мире долгое время являлся и продолжает оставаться метод кариотипирования с дифференциальной окраской хромосом. Этот метод позволяет анализировать кариотип в целом и определять крупные (не менее 5-10 млн пар нуклеотидов) хромосомные перестройки. Однако у него существует ряд ограничений, таких как трудоемкость, длительность (1-2 недели), высокие требования к квалификации и опыту специалиста, проводящего исследование, а также, в ряде случаев, технические проблемы (недостаточное количество и качество исследуемого материала, отсутствие митозов или роста культуры).
Этих недостатков лишен метод количественной флуоресцентной полимеразной цепной реакции (КФ-ПЦР), который все более широко применяется для диагностики анеуплоидий, в том числе и синдрома Эдвардса (Рис. 1). Этот метод обладает достоверностью, сравнимой с достоверностью стандартного кариотипирования, является более быстрым, дешевым, менее требовательным к количеству и качеству материала (поскольку не связан с ростом культуры клеток) и позволяет одновременно анализировать большое число образцов. Однако метод КФ-ПЦР имеет и ограничения: в мозаичных случаях он позволяет выявлять только высокоуровневый мозаицизм (от 20%), кроме того, он не может исключить наличие более редких хромосомных нарушений, которые могут быть связаны с пороками развития плода. При проведении дородовой диагностики синдрома Эдвардса, кроме материала плода, необходимо предоставлять биологический материал матери для того, чтобы исключить возможность получения ложноотрицательного результата из-за неправильного забора плодного материала. Анализ плодного материала выполняется за три рабочих дня.
При синдроме Эдвардса отмечается выраженная задержка пренатального развития, дети рождаются с пренатальной гипотрофией (средняя масса тела при рождении составляет 2340 г). Внешние проявления синдрома Эдвардса многообразны (Рис. 2). Наиболее типичными являются задержка психомоторного развития, гипоплазия скелетной мускулатуры и подкожной жировой ткани, врожденные пороки сердца, аномалии строения лица и черепа (долихоцефалия, микрофтальмия, укорочение глазных щелей, низкое расположение ушных раковин, микрогнатия, скошенный подбородок), множественные деформации кистей и стоп, аномалии развития желудочно-кишечного тракта, мочеполовой системы и центральной нервной системы (спинно-мозговые грыжи, гипоплазия мозолистого тела и мозжечка). Продолжительность жизни детей резко снижена: 90% из них погибают до года от осложнений, обусловленных врождёнными пороками развития (асфиксия, пневмония, кишечная непроходимость, сердечно-сосудистая недостаточность).
Причиной развития синдрома Эдвардса является утроение хромосомы 18. Трисомия по хромосоме 18 является частным случаем анеуплоидии– наличия в геноме набора хромосом, отличного от стандартного для данного вида и некратного ему. Трисомия хромосомы 18 обычно вызвана нерасхождением хромосом при формировании половых клеток родителя (яйцеклеток и сперматозоидов), в результате чего ребенок получает от матери или от отца лишнюю 18-ю хромосому. В этом случае все клетки организма ребёнка будут нести аномалию. В том случае, когда нерасхождение хромосом возникает при делении какой-либо клетки зародыша, наблюдается мозаичный вариант синдрома Эдвардса (10% случаев).
Риск рождения детей с синдромом Эдвардса, по разным литературным данным, не изменяется или незначительно возрастает с увеличением возраста беременной женщины.
Пренатальная диагностика синдрома Эдвардса включает в себя два этапа. На первом этапе, на сроке беременности 11-13 недель, проводится скрининг, который основывается преимущественно на биохимических показателях, поскольку на ранних сроках УЗИ не позволяет обнаружить в случае синдрома Эдвардса каких-либо грубых аномалий развития, которые могут быть выявлены лишь к 20-24 неделе. Биохимический анализ уровня определенных белков в крови беременной женщины (свободной β-субъединицы хорионического гормона человека (β-ХГЧ) и ассоциированного с беременностью плазменного протеина А (pregnancy associated plasma protein-A, РАРР-А)), с учетом ее возраста, позволяет рассчитать для нее риск рождения больного ребенка. Однако эти методы не позволяют поставить точный диагноз, и в результате проведенного скрининга лишь формируется группа риска беременных с повышенной вероятностью рождения больного синдромом Эдвардса. На втором этапе в группе риска проводится инвазивная процедура для получения плодного материала, необходимого для точного определения статуса плода. В зависимости от срока беременности это может быть биопсия ворсин хориона (8-12 недели), амниоцентез (14-18 недели) или кордоцентез (после 20-й недели). В полученных образцах ткани плода проводится определение хромосомного набора.
В Центре Молекулярной Генетики проводится диагностика синдрома Эдвардса (в том числе и пренатальная) методом КФ-ПЦР.
Синдром Эдвардса (трисомия 18). Причины, симптомы, признаки, диагностика и лечение патологии
Сайт предоставляет справочную информацию. Адекватная диагностика и лечение болезни возможны под наблюдением добросовестного врача.
 Синдром Эдвардса
или трисомия 18
представляет собой тяжелое врожденное заболевание, вызванное хромосомными нарушениями. Оно является одной из наиболее распространенных патологий в данной категории (уступает по частоте лишь синдрому Дауна
). Заболевание характеризуется многочисленными нарушениями в развитии различных органов и систем. Прогноз для ребенка обычно неблагоприятный, но многое зависит от ухода, который ему способны обеспечить родители.
Синдром Эдвардса
или трисомия 18
представляет собой тяжелое врожденное заболевание, вызванное хромосомными нарушениями. Оно является одной из наиболее распространенных патологий в данной категории (уступает по частоте лишь синдрому Дауна
). Заболевание характеризуется многочисленными нарушениями в развитии различных органов и систем. Прогноз для ребенка обычно неблагоприятный, но многое зависит от ухода, который ему способны обеспечить родители.
Распространенность синдрома Эдвардса по земному шару варьирует от 0,015 до 0,02%. Четкой зависимости от местности или расы не наблюдается. Статистически девочки болеют в 3 – 4 раза чаще мальчиков. Научного объяснения этой пропорции пока не выявлено. Тем не менее, отмечен ряд факторов, которые могут повысить риск возникновения этой патологии.
Как и другие хромосомные мутации, синдром Эдвардса является, в принципе, неизлечимым заболеванием. Самые современные методы лечения и ухода могут лишь поддерживать жизнь ребенка и способствовать определенному прогрессу в его развитии. Единых рекомендаций по уходу за такими детьми нет из-за огромного разнообразия возможных нарушений и осложнений.
Интересные факты
- Описание основных симптомов данной болезни было сделано еще в начале XX века.
- До середины 1900-х годов собрать достаточную информацию об этой патологи не представлялось возможным. Во-первых, для этого необходим был соответствующий уровень развития технологий, который позволил бы обнаружить лишнюю хромосому. Во-вторых, большинство детей умирало в первые дни или недели жизни из-за низкого уровня оказания медицинской помощи.
- Первое полное описание болезни и ее основной причины (появление лишней 18-й хромосомы ) было сделано только в 1960 году врачом Джоном Эдвардом, в честь которого тогда и назвали новую патологию.
- Реальная частота синдрома Эдвардса составляет 1 случай на 2,5 – 3 тысячи зачатий (0,03 – 0,04% ), однако официальные данные значительно ниже. Это объясняется тем, что почти половина эмбрионов с данной аномалией не выживают и беременность заканчивается спонтанным абортом или внутриутробной смертью плода. Подробная диагностика причины выкидыша при этом проводится редко.
- Трисомия представляет собой вариант хромосомной мутации, при которой у человека в клетках содержится не 46, а 47 хромосом. Существует всего 3 синдрома в данной группе заболеваний. Помимо синдрома Эдвардса это синдромы Дауна (трисомия 21 хромосомы ) и Патау (трисомия 13 хромосомы ). При наличии других добавочных хромосом патология несовместима с жизнью. Лишь в этих трех случаях возможно рождение живого ребенка и его дальнейший (хоть и замедленный ) рост и развитие.
Причины генетической патологии
 Синдром Эдвардса является генетическим заболеванием
, которое характеризуется наличием дополнительной хромосомы в геноме человека. Чтобы разобраться в причинах, которые вызывают видимые проявления этой патологии, необходимо выяснить, что представляют собой собственно хромосомы и генетический материал в целом.
Синдром Эдвардса является генетическим заболеванием
, которое характеризуется наличием дополнительной хромосомы в геноме человека. Чтобы разобраться в причинах, которые вызывают видимые проявления этой патологии, необходимо выяснить, что представляют собой собственно хромосомы и генетический материал в целом.
В каждой клетке человека имеется ядро, которое отвечает за хранение и обработку генетической информации. В ядре содержится 46 хромосом (23 пары ), которые представляют собой многократно упакованную молекулу ДНК (дезоксирибонуклеиновая кислота ). Эта молекула содержит в себе определенные участки, называемые генами. Каждый ген является прототипом определенного белка в организме человека. При необходимости клетка считывает информацию с этого прототипа и вырабатывает соответствующий белок. Дефекты генов ведут к производству аномальных белков, которые и ответственны за появление генетических заболеваний.
Хромосомная пара состоит из двух идентичных молекул ДНК (одна отцовская, другая – материнская ), которые сцеплены между собой небольшим мостиком (центромерой ). Место сцепления двух хромосом в паре предопределяет форму всего соединения и его вид под микроскопом.
Все хромосомы хранят различную генетическую информацию (о разных белках) и подразделяются на следующие группы:
- группа А включает 1 – 3 пару хромосом, которые отличаются большими размерами и X-образной формой;
- группа В включает 4 – 5 пару хромосом, которые также являются крупными, но центромера лежит дальше от центра, из-за чего форма напоминает букву Х со смещенным вниз или вверх центром;
- группа С включает 6 – 12 пару хромосом, которые по форме напоминают хромосомы группы В, но уступают им по размерам;
- группа D включает 13 – 15 пару хромосом, для которых характерны средние размеры и расположение центромеры у самого конца молекул, что придает сходство с буквой V;
- группа Е включает 16 – 18 пару хромосом, которые характеризуются маленькими размерами и срединным расположением центромеры (форма буквы Х );
- группа F включает 19 – 20 хромосомные пары, которые несколько мельче хромосом группы Е и схожи с ними по форме;
- группа G включает 21 – 22 пару хромосом, которые характеризуются V-образной формой и очень маленькими размерами.
Вышеперечисленные 22 пары хромосом называются соматическими или аутосомами. Кроме того существуют половые хромосомы, которые и составляют 23 пару. Они не похожи по внешнему виду, поэтому каждая из них обозначается отдельно. Женская половая хромосома обозначается Х и сходна с группой С. Мужская половая хромосома обозначается Y и сходна по форме и размерам с группой G. Если у ребенка обе хромосомы женские (тип ХХ ), то рождается девочка. Если же одна из половых хромосом женская, а другая мужская, то рождается мальчик (тип ХY ). Хромосомная формула называется кариотипом и может быть обозначена следующим образом — 46,ХХ. Здесь число 46 обозначает общее количество хромосом (23 пары ), а ХХ – формулу половых хромосом, которая зависит от пола (в примере представлен кариотип нормальной женщины ).
Синдром Эдвардса относится к так называемым хромосомным заболеваниям, когда проблема состоит не в дефекте гена, а в дефекте целой молекулы ДНК. Если быть более точным, то классическая форма этого заболевания подразумевает наличие лишней 18-й хромосомы. Кариотип в таких случаях обозначается как 47,ХХ, 18+ (для девочки ) и 47,ХY, 18+ (для мальчика ). Последняя цифра обозначает номер добавочной хромосомы. Излишек генетической информации в клетках приводит к появлению соответствующих проявлений болезни, которые и объединены под названием «синдром Эдвардса». Наличие дополнительной (третьей ) хромосомы под номером 18 дало другое (более научное ) название болезни – трисомия 18.
В зависимости от формы хромосомного дефекта различают три вида данного заболевания:
- Полная трисомия 18 . Полная или классическая форма синдрома Эдвардса предполагает, что все клетки в организме имеют дополнительную хромосому. Данный вариант заболевания встречается более чем в 90% случаев и является наиболее тяжелым.
- Частичная трисомия 18 . Частичная трисомия 18 является весьма редким феноменом (не более 3% от всех случаев синдрома Эдвардса ). При ней в клетках организма содержится не целая дополнительная хромосома, а лишь ее фрагмент. Такой дефект может быть результатом неправильного деления генетического материала, но встречается он очень редко. Иногда часть восемнадцатой хромосомы присоединяется к другой молекуле ДНК (внедряется в ее структуру, удлиняя молекулу, или просто «цепляется» с помощью мостика ). Последующее деление клеток приводит к тому, что в организме имеется 2 нормальные хромосомы номер 18 и еще часть генов с этих хромосом (сохранившийся фрагмент молекулы ДНК ). В этом случае количество врожденных дефектов будет намного ниже. Наблюдается избыток не всей генетической информации, закодированной в 18-й хромосоме, а лишь ее части. Для пациентов с частичной трисомией 18 прогноз лучше, чем для детей с полной формой, но все равно остается неблагоприятным.
- Мозаичная форма . Мозаичная форма синдрома Эдвардса встречается в 5 – 7% случаев данного заболевания. Механизм ее появления отличается от других видов. Дело в том, что здесь дефект образовался уже после слияния сперматозоида и яйцеклетки. Обе гаметы (половые клетки ) изначально имели нормальный кариотип и несли по одной хромосоме каждого вида. После слияния сформировалась клетка с нормальной формулой 46,ХХ или 46,XY. В процессе деления этой клетки произошел сбой. При удвоении генетического материала один из фрагментов получил дополнительную 18-ю хромосому. Таким образом, на определенном этапе сформировался зародыш, часть клеток которого имеют нормальный кариотип (например, 46,ХХ ), а часть – кариотип синдрома Эдвардса (47,ХХ, 18+ ). Доля патологических клеток никогда не превышает 50%. Их число зависит от того, на каком этапе деления начальной клетки произошел сбой. Чем позже это происходит, тем меньше будет доля дефектных клеток. Форма получила название из-за того, что все клетки организма представляют собой своеобразную мозаику. Часть из них здорова, а часть – с тяжелой генетической патологией. Закономерности в распределении клеток в организме при этом не наблюдается, то есть все дефектные клетки не могут локализоваться только в одном месте, чтобы их можно было удалить. Общее состояние пациента при этом легче, чем при классической форме трисомии 18.
Наличие дополнительной хромосомы в геноме человека представляет множество проблем. Дело в том, что клетки человека запрограммированы считывать генетическую информацию и дублировать лишь заданное природой количество молекул ДНК. Нарушения даже в структуре одного гена могут привести к серьезным заболеваниям. При наличии же целой молекулы ДНК развиваются множественные нарушения еще на этапе внутриутробного развития до рождения ребенка.
Согласно последним исследованиям хромосома номер 18 содержит 557 генов, которые кодируют не менее 289 различных белков. В процентном отношении это примерно 2,5% всего генетического материала. Нарушения, которые вызывает столь большой дисбаланс, очень серьезны. Неправильное количество белков предопределяет множество аномалий в развитии различных органов и тканей. В случае синдрома Эдвардса чаще других страдают кости черепа, некоторые отделы нервной системы, сердечно-сосудистая и мочеполовая система. По всей видимости, это связано с тем, что гены, расположенные на этой хромосоме, имеют отношение к развитию именно этих органов и систем.
Таким образом, основной и единственной причиной синдрома Эдвардса является наличие дополнительной молекулы ДНК. Наиболее часто (при классической форме болезни ) она наследуется от одного из родителей. В норме каждая гамета (сперматозоид и яйцеклетка ) содержат по 22 непарные соматические хромосомы, плюс одна половая. Женщина всегда передает ребенку стандартный набор 22+Х, а мужчина может передать 22+Х либо 22+Y. Это предопределяет пол ребенка. Половые клетки родителей образуются в результате разделения обычных клеток на два набора. В норме материнская клетка делится на две равные части, но иногда не все хромосомы делятся пополам. Если 18-я пара не разошлась по полюсам клетки, то одна из яйцеклеток (или одни из сперматозоидов ) будет заранее дефектным. В нем будет не 23, а 24 хромосомы. В случае если именно эта клетка будет участвовать в оплодотворении, ребенок получит дополнительную 18-ю хромосому.
На неправильное деление клеток могут повлиять следующие факторы:
- Возраст родителей . Доказано, что вероятность хромосомных аномалий увеличивается прямо пропорционально с возрастом матери. При синдроме Эдвардса эта связь менее выражена, чем при других похожих патологиях (например, синдром Дауна ). Но для женщин старше 40 лет риск родить ребенка с данной патологией в среднем в 6 – 7 раз выше. Подобная зависимость от возраста отца наблюдается в значительно меньшей степени.
- Курение и алкоголь . Такие вредные привычки как курение и злоупотребление алкоголем могут действовать на половую систему человека, влияя на деление половых клеток. Таким образом, регулярное употребление этих веществ (а также других наркотических препаратов ) повышает риск неправильного распределения генетического материала.
- Прием лекарственных средств . Некоторые лекарственные препараты при неправильном приеме в первом триместре могут повлиять на деление зародышевых клеток и спровоцировать мозаичную форму синдрома Эдвардса.
- Заболевания половой сферы. Перенесенные инфекции с поражением репродуктивных органов могут отразиться на правильном делении клеток. Они повышают риск хромосомных и генетических заболеваний в целом, хотя специально для синдрома Эдвардса подобные исследования не проводились.
- Радиационное излучение. Облучение половых органов рентгеновским излучением или другими ионизирующими излучениями может повлечь генетические мутации. Особенно опасно такое внешнее воздействие в подростковом возрасте, когда деление клеток происходит наиболее активно. Частицы, формирующие излучение, легко проникают сквозь ткани и подвергают молекулу ДНК своеобразной «бомбардировке». Если это происходит в момент деления клетки, риск хромосомной мутации особенно высок.
В целом же нельзя сказать, что причины развития синдрома Эдвардса окончательно известны и хорошо изучены. Вышеперечисленные факторы лишь повышают риск развития данной мутации. Не исключается и врожденная предрасположенность некоторых людей к неправильному распределению генетического материала в половых клетках. Например, считается, что у супружеской пары, которая уже родила ребенка с синдромом Эдвардса, вероятность появления второго ребенка с аналогичной патологией составляет аж 2 – 3% (примерно в 200 раз выше, чем средняя распространенность этой болезни ).
Как выглядят новорожденные с синдромом Эдвардса?
 Как известно, диагностировать синдром Эдвардса можно до рождения, но в большинстве случаев данное заболевание обнаруживают непосредственно после рождения ребенка. У новорожденных с данной патологией имеется ряд ярко выраженных аномалий развития, которые иногда позволяют сразу заподозрить правильный диагноз. Подтверждение проводится позже при помощи специального генетического анализа.
Как известно, диагностировать синдром Эдвардса можно до рождения, но в большинстве случаев данное заболевание обнаруживают непосредственно после рождения ребенка. У новорожденных с данной патологией имеется ряд ярко выраженных аномалий развития, которые иногда позволяют сразу заподозрить правильный диагноз. Подтверждение проводится позже при помощи специального генетического анализа.
Новорожденные с синдромом Эдвардса имеют следующие характерные аномалии развития:
- изменение формы черепа;
- изменение формы ушных раковин;
- аномалии развития неба;
- стопа-качалка;
- аномальная длина пальцев;
- изменение формы нижней челюсти;
- сращение пальцев;
- аномалии развития половых органов;
- флексорное положение кистей;
- дерматоглифические признаки.
Изменение формы черепа
 Типичным симптомом при синдроме Эдвардса является долихоцефалия. Так называется характерное изменение формы головы новорожденного ребенка, которое встречается и при некоторых других генетических заболеваниях. У долихоцефалов (детей с данным симптомом
) более длинный и узкий череп. Точно подтверждается наличие этой аномалии с помощью специальных измерений. Определяют соотношение ширины черепа на уровне теменных костей к длине черепа (от выступа над переносицей до затылочного бугра
). Если полученное соотношение меньше 75%, то данный ребенок относится к долихоцефалам. Сам по себе данный симптом не является серьезным нарушением. Это просто один из видов формы черепа, встречающийся и у абсолютно нормальных людей. Дети с синдромом Эдвардса в 80 – 85% случаев являются выраженными долихоцефалами, у которых диспропорцию длины и ширины черепа можно заметить и без специальных измерений.
Типичным симптомом при синдроме Эдвардса является долихоцефалия. Так называется характерное изменение формы головы новорожденного ребенка, которое встречается и при некоторых других генетических заболеваниях. У долихоцефалов (детей с данным симптомом
) более длинный и узкий череп. Точно подтверждается наличие этой аномалии с помощью специальных измерений. Определяют соотношение ширины черепа на уровне теменных костей к длине черепа (от выступа над переносицей до затылочного бугра
). Если полученное соотношение меньше 75%, то данный ребенок относится к долихоцефалам. Сам по себе данный симптом не является серьезным нарушением. Это просто один из видов формы черепа, встречающийся и у абсолютно нормальных людей. Дети с синдромом Эдвардса в 80 – 85% случаев являются выраженными долихоцефалами, у которых диспропорцию длины и ширины черепа можно заметить и без специальных измерений.
Другим вариантом аномалии развития черепа является так называемая микроцефалия, при которой размеры головы в целом слишком малы по сравнению с остальным туловищем. Прежде всего, это касается не лицевого черепа (челюсти, скулы, глазницы ), а именно черепной коробки, в которой располагается мозг. Микроцефалия менее характерна для синдрома Эдвардса, чем долихоцефалия, но она тоже встречается с более высокой частотой, чем среди здоровых людей.
Изменение формы ушной раковины
 Если долихоцефалия может быть вариантом нормы, то патологии развития ушной раковины у детей с синдромом Эдвардса куда тяжелее. В определенной степени этот симптом наблюдается более чем у 95% детей с полной формой данного заболевания. При мозаичной форме его частота несколько меньше. Ушная раковина обычно располагается ниже, чем у нормальных людей (иногда ниже уровня глаз
). Характерные выпуклости хряща, который формирует ушную раковину, плохо выражены или отсутствуют. Также могут отсутствовать мочка или козелок (небольшой выступающий участок хряща спереди от слухового отверстия
). Сам слуховой проход обычно сужен, а примерно в 20 – 25% — вовсе отсутствует.
Если долихоцефалия может быть вариантом нормы, то патологии развития ушной раковины у детей с синдромом Эдвардса куда тяжелее. В определенной степени этот симптом наблюдается более чем у 95% детей с полной формой данного заболевания. При мозаичной форме его частота несколько меньше. Ушная раковина обычно располагается ниже, чем у нормальных людей (иногда ниже уровня глаз
). Характерные выпуклости хряща, который формирует ушную раковину, плохо выражены или отсутствуют. Также могут отсутствовать мочка или козелок (небольшой выступающий участок хряща спереди от слухового отверстия
). Сам слуховой проход обычно сужен, а примерно в 20 – 25% — вовсе отсутствует.
Аномалии развития неба
 Небные отростки верхней челюсти в процессе развития эмбриона срастаются, формируя твердое небо. У детей с синдромом Эдвардса этот процесс нередко остается незавершенным. В том месте, где у нормальных людей располагается срединный шов (его можно прощупать посередине твердого неба языком
) у них идет продольная щель.
Небные отростки верхней челюсти в процессе развития эмбриона срастаются, формируя твердое небо. У детей с синдромом Эдвардса этот процесс нередко остается незавершенным. В том месте, где у нормальных людей располагается срединный шов (его можно прощупать посередине твердого неба языком
) у них идет продольная щель.
Существует несколько вариантов данного дефекта:
- незаращение мягкого неба (задняя, глубокая часть неба, которая нависает над глоткой );
- частичное незаращение твердого неба (щель не тянется на протяжении всей верхней челюсти );
- полное незаращение твердого и мягкого неба;
- полное незаращение неба и губы.
В ряде случаев расщепление неба является двусторонним. Два выступающих вверх уголка верхней губы являются началом патологических щелей. Ребенок не может полностью закрыть рот из-за этого дефекта. В тяжелых случаях явно видно сообщение ротовой и носовой полости (даже при закрытом рте ). Передние зубы в будущем могут отсутствовать или расти в сторону.
Данные дефекты развития известны также под названием волчья пасть, расщепление неба, заячья губа. Все они могут встречаться и не в рамках синдрома Эдвардса, однако у детей с этой патологией их частота особенно высока (почти 20% новорожденных ). Значительно чаще (до 65% новорожденных ) обладают другой особенностью, известной как высокое или готическое небо. Оно может быть отнесено к вариантам нормы, так как встречается и у здоровых людей.
Наличие расщепленного верхнего неба или верхней губы еще не подтверждает синдром Эдвардса. Этот порок развития может встречаться с довольно высокой частотой и самостоятельно без сопутствующих нарушений со стороны других органов и систем. Для исправления данной аномалии существует ряд стандартных хирургических вмешательств.
Стопа-качалка
 Так называется характерное изменение стопы, которое встречается, в основном, в рамках синдрома Эдвардса. Частота его при данном заболевании достигает 75%. Дефект заключается в неправильном взаимоположении таранной, пяточной и ладьевидной костей. Его относят к категории плоско-вальгусных деформаций стопы у детей.
Так называется характерное изменение стопы, которое встречается, в основном, в рамках синдрома Эдвардса. Частота его при данном заболевании достигает 75%. Дефект заключается в неправильном взаимоположении таранной, пяточной и ладьевидной костей. Его относят к категории плоско-вальгусных деформаций стопы у детей.
Внешне стопа у новорожденного ребенка выглядит следующим образом. Пяточный бугор, на который опирается задняя часть стопы, выдается назад. Свод при этом может полностью отсутствовать. Это легко заметить, посмотрев на стопу с внутренней стороны. В норме там вырисовывается вогнутая линия, направляющаяся от пятки к основанию большого пальца. При стопе-качалке этой линии нет. Стопа плоская или даже выпуклая. Это и придает ей сходство с ножками кресла-качалки.
Аномальная длина пальцев
 У детей с синдромом Эдвардса на фоне изменений в строении стопы может наблюдаться ненормальная пропорция в длине пальцев ног. В частности, речь идет о большом пальце, который в норме является самым длинным. У новорожденных с данным синдромом он уступает по длине второму пальцу. Данный дефект можно заметить лишь при распрямлении пальцев и тщательном их рассмотрении. С возрастом, по мере роста ребенка, он становится более заметным. Поскольку укорочение большого пальца стопы встречается в основном при стопе-качалке, распространенность этих симптомов у новорожденных примерно одинакова.
У детей с синдромом Эдвардса на фоне изменений в строении стопы может наблюдаться ненормальная пропорция в длине пальцев ног. В частности, речь идет о большом пальце, который в норме является самым длинным. У новорожденных с данным синдромом он уступает по длине второму пальцу. Данный дефект можно заметить лишь при распрямлении пальцев и тщательном их рассмотрении. С возрастом, по мере роста ребенка, он становится более заметным. Поскольку укорочение большого пальца стопы встречается в основном при стопе-качалке, распространенность этих симптомов у новорожденных примерно одинакова.
У взрослых укорочение большого пальца на ноге не имеет такой диагностической ценности. Подобный дефект может быть индивидуальной особенностью у здорового человека или следствием воздействия других факторов (деформация суставов, заболевания костей, ношение обуви, не соответствующей по размеру ). В связи с этим данный признак нужно рассматривать как возможный симптом только у новорожденных детей при наличии других аномалий развития.
Изменение формы нижней челюсти
 Изменения формы нижней челюсти у новорожденных встречаются почти в 70% случаев. В норме подбородок у детей не выступает вперед так, как у взрослых, но у больных с синдромом Эдвардса он слишком уж сильно втянут. Это происходит из-за недоразвития нижней челюсти, которое носит название микрогнатия (микрогения
). Данный симптом встречается и при других врожденных заболеваниях. Не так уж редко можно встретить и взрослых людей с похожими чертами лица. При отсутствии сопутствующих патологий это считается вариантом нормы, хоть и ведет к некоторым трудностям.
Изменения формы нижней челюсти у новорожденных встречаются почти в 70% случаев. В норме подбородок у детей не выступает вперед так, как у взрослых, но у больных с синдромом Эдвардса он слишком уж сильно втянут. Это происходит из-за недоразвития нижней челюсти, которое носит название микрогнатия (микрогения
). Данный симптом встречается и при других врожденных заболеваниях. Не так уж редко можно встретить и взрослых людей с похожими чертами лица. При отсутствии сопутствующих патологий это считается вариантом нормы, хоть и ведет к некоторым трудностям.
У новорожденных с микрогнатией обычно быстро появляются следующие проблемы:
- невозможность долго держать рот закрытым (подтекание слюны );
- затруднения при кормлении;
- позднее развитие зубов и неправильное их расположение.
Зазор между нижней и верхней челюстью может составлять более 1 см, что очень много, учитывая размеры головы малыша.
Сращение пальцев
 Сращение пальцев, или по-научному синдактилия, наблюдается приблизительно у 45% новорожденных. Чаще всего эта аномалия затрагивает пальцы ног, но встречается и синдактилия на руках. В легких случаях сращение образовано кожной складкой наподобие короткой перепонки. В более тяжелых случаях наблюдается сращение мостиками костной ткани.
Сращение пальцев, или по-научному синдактилия, наблюдается приблизительно у 45% новорожденных. Чаще всего эта аномалия затрагивает пальцы ног, но встречается и синдактилия на руках. В легких случаях сращение образовано кожной складкой наподобие короткой перепонки. В более тяжелых случаях наблюдается сращение мостиками костной ткани.
Синдактилия встречается не только при синдроме Эдвардса, но и при многих других хромосомных заболеваниях. Известны и случаи, когда этот порок развития являлся единственным, и в остальном больной ничем не отличался от нормальных детей. В связи с этим сращение пальцев является лишь одним из возможных признаков синдрома Эдвардса, который помогает заподозрить диагноз, но не подтверждает его.
Аномалии развития половых органов
Флексорное положение кистей
 Флексорное положение кистей – это особое расположение пальцев, вызванное не столько структурными нарушениями в области кисти, сколько повышенным тонусом мышц. Сгибатели пальцев и кисти постоянно напряжены, из-за чего большой палец и мизинец как бы прикрывают остальные пальцы, которые при этом прижаты к ладони. Данный симптом наблюдается при многих врожденных патологиях и не является характерным именно для синдрома Эдвардса. Тем не менее, при обнаружении кисти подобной формы необходимо предполагать эту патологию. При ней флексорное положение пальцев наблюдается почти у 90% новорожденных.
Флексорное положение кистей – это особое расположение пальцев, вызванное не столько структурными нарушениями в области кисти, сколько повышенным тонусом мышц. Сгибатели пальцев и кисти постоянно напряжены, из-за чего большой палец и мизинец как бы прикрывают остальные пальцы, которые при этом прижаты к ладони. Данный симптом наблюдается при многих врожденных патологиях и не является характерным именно для синдрома Эдвардса. Тем не менее, при обнаружении кисти подобной формы необходимо предполагать эту патологию. При ней флексорное положение пальцев наблюдается почти у 90% новорожденных.
Дерматоглифические признаки
 При многих хромосомных аномалиях у новорожденных имеются характерные дерматоглифические изменения (аномальные узоры и складки на коже ладоней
). При синдроме Эдвардса некоторые признаки можно обнаружить почти в 60% случаев. Они имеют значение в основном для предварительной диагностики при мозаичной или частичной форме болезни. При полной трисомии 18 к дерматоглифике не прибегают, так как для подозрения синдрома Эдвардса хватает других, более заметных аномалий развития.
При многих хромосомных аномалиях у новорожденных имеются характерные дерматоглифические изменения (аномальные узоры и складки на коже ладоней
). При синдроме Эдвардса некоторые признаки можно обнаружить почти в 60% случаев. Они имеют значение в основном для предварительной диагностики при мозаичной или частичной форме болезни. При полной трисомии 18 к дерматоглифике не прибегают, так как для подозрения синдрома Эдвардса хватает других, более заметных аномалий развития.
Основными дерматоглифическими признаками синдрома Эдвардса являются:
- дуги на подушечках пальцев располагаются с большей частотой, нежели у здоровых людей;
- кожная складка между последней (ногтевой ) и предпоследней (срединной ) фалангами пальцев отсутствует;
- у 30% новорожденных на ладони имеется так называемая поперечная борозда (обезьянья линия, линия Симиан ).
Специальные исследования могут выявить и другие отклонения от нормы, однако непосредственно после рождения, без привлечения узких специалистов, врачам достаточно этих изменений.
Помимо вышеперечисленных признаков существует еще целый ряд возможных аномалий развития, которые могут помочь в предварительной диагностике синдрома Эдвардса. По некоторым данными при подробном внешнем осмотре можно обнаружить до 50 внешних признаков. Сочетание наиболее частых симптомов, представленных выше, с высокой вероятностью говорит о наличии у ребенка этой тяжелой патологии. При мозаичном варианте синдрома Эдвардса множественных аномалий может и не быть, однако наличие даже одного из них является показанием к проведению специального генетического теста.
Как выглядят дети с синдромом Эдвардса?
 У детей с синдромом Эдвардса по мере взросления обычно обнаруживаются самые разные сопутствующие патологии. Их симптомы начинают проявляться уже через несколько недель после рождения. Эти симптомы могут оказаться первым проявлением синдрома, так как при мозаичном варианте в редких случаях болезнь может остаться незамеченной непосредственно после рождения. Тогда диагностика заболевания усложняется.
У детей с синдромом Эдвардса по мере взросления обычно обнаруживаются самые разные сопутствующие патологии. Их симптомы начинают проявляться уже через несколько недель после рождения. Эти симптомы могут оказаться первым проявлением синдрома, так как при мозаичном варианте в редких случаях болезнь может остаться незамеченной непосредственно после рождения. Тогда диагностика заболевания усложняется.
Большинство внешних проявлений синдрома, замеченных при рождении, остаются и становятся более заметными. Речь идет о форме черепа, стопе-качалке, деформации ушной раковины и т. п. Постепенно к ним начинают прибавляться и другие внешние проявления, которые невозможно было заметить сразу после рождения. В данном случае речь идет о признаках, которые могут появиться у детей в первый год жизни.
Дети с синдромом Эдвардса имеют следующие внешние особенности:
- отставание в физическом развитии;
- косолапость;
- аномальный тонус мышц;
- ненормальные эмоциональные реакции.
Отставание в физическом развитии
Отставание в физическом развитии объясняется низкой массой тела ребенка при рождении (всего 2000 – 2200 г при нормальном сроке беременности ). Значительную роль играет и генетический дефект, который не позволяет всем системам организма нормально и гармонично развиваться. Основные показатели, по которым оценивают рост и развитие ребенка, сильно снижены.
Заметить отставание ребенка можно по следующим антропометрическим показателям:
- рост ребенка;
- вес ребенка;
- окружность грудной клетки;
- окружность головы (данный показатель может быть в норме или даже увеличен, но на него нельзя полагаться из-за врожденной деформации черепа ).
Косолапость
Аномальный тонус мышц
Ненормальные эмоциональные реакции
Как выглядят взрослые с синдромом Эдвардса?
 В подавляющем большинстве случаев дети, рожденные с синдромом Эдвардса, не доживают до взрослого возраста. При полной форме этого заболевания, когда лишняя хромосома присутствует в каждой клетке тела, 90% детей умирает в возрасте до 1 года из-за серьезных аномалий развития внутренних органов. Даже при условии хирургического исправления возможных дефектов и качественном уходе их организм более подвержен инфекционным заболеваниям. Этому способствуют и нарушения питания, которые встречаются у большинства детей. Все это объясняет высочайшую смертность при синдроме Эдвардса.
В подавляющем большинстве случаев дети, рожденные с синдромом Эдвардса, не доживают до взрослого возраста. При полной форме этого заболевания, когда лишняя хромосома присутствует в каждой клетке тела, 90% детей умирает в возрасте до 1 года из-за серьезных аномалий развития внутренних органов. Даже при условии хирургического исправления возможных дефектов и качественном уходе их организм более подвержен инфекционным заболеваниям. Этому способствуют и нарушения питания, которые встречаются у большинства детей. Все это объясняет высочайшую смертность при синдроме Эдвардса.
При более легкой мозаичной форме, когда лишь часть клеток в организме содержит аномальный набор хромосом, выживаемость несколько больше. Однако даже в этих случаях до взрослого возраста доживают единичные пациенты. Их внешний вид определяется врожденными аномалиями, которые присутствовали при рождении (заячья губа, деформированная ушная раковина, и др. ). Основным же симптомом, присутствующим у всех без исключения детей, является серьезнейшее отставание в умственном развитии. Дожив до взрослого возраста, ребенок с синдромом Эдвардса является глубоким олигофреном (IQ менее 20, что соответствует самой тяжелой степени умственной отсталости ). В целом же в медицинской литературе описываются единичные случаи, когда дети с синдромом Эдвардса доживали до совершеннолетнего возраста. Из-за этого накоплено слишком мало объективных данных, чтобы говорить о внешних признаках этого заболевания у взрослых.
Диагностика генетической патологии
 В настоящее время существуют три основных этапа диагностики синдрома Эдвардса, каждый из которых включает несколько возможных методов. Поскольку данное заболевание является неизлечимым, родителям следует обратить внимание на возможности этих методов и воспользоваться ими. Большинство анализов проводится в специальных центрах пренатальной диагностики, где имеется вся необходимая техника для поиска генетических заболеваний. Однако даже консультация у врача-генетика или неонатолога может оказаться полезной.
В настоящее время существуют три основных этапа диагностики синдрома Эдвардса, каждый из которых включает несколько возможных методов. Поскольку данное заболевание является неизлечимым, родителям следует обратить внимание на возможности этих методов и воспользоваться ими. Большинство анализов проводится в специальных центрах пренатальной диагностики, где имеется вся необходимая техника для поиска генетических заболеваний. Однако даже консультация у врача-генетика или неонатолога может оказаться полезной.
Диагностика синдрома Эдвардса возможна на следующих этапах:
- диагностика до момента зачатия;
- диагностика во время внутриутробного развития;
- диагностика после рождения.
Диагностика до момента зачатия
Диагностика до момента зачатия ребенка является идеальным вариантом, но, к сожалению, на современном этапе развития медицины ее возможности очень ограничены. Врачи могут с помощью нескольких методов предположить повышенную вероятность рождения ребенка с хромосомным заболеванием, но не более того. Дело в том, что при синдроме Эдвардса, в принципе, нарушения у родителей обнаружить нельзя. Дефектная половая клетка с 24 хромосомами является лишь одной из многих тысяч. Поэтому сказать наверняка до момента зачатия, родится ли ребенок с данным заболеванием, нельзя.
Основными методами диагностики до момента зачатия являются:
- Семейный анамнез . Семейный анамнез представляет собой подробный опрос обоих родителей об их родословной. Врача интересуют любые случаи наследственных (и особенно хромосомных ) заболеваний в семье. Если хотя бы один из родителей припоминает случай трисомии (синдром Эдвардса, Дауна, Патау ), это сильно повышает вероятность рождения больного ребенка. Однако риск все равно составляет не более 1%. При повторных случаях этих заболеваний у предков риск многократно возрастает. По сути, анализ сводится к консультации у неонатолога или генетика. Предварительно родители могут постараться собрать более подробную информацию о своих предках (желательно на 3 – 4 колена ). Это повысит точность данного метода.
- Обнаружение факторов риска . Основным фактором риска, который объективно повышает риск хромосомных аномалий, является возраст матери. Как уже говорилось выше, у матерей после 40 лет вероятность рождения ребенка с синдромом Эдвардса многократно возрастает. По некоторым данным, после 45 лет (возраст матери ) почти каждая пятая беременность сопровождается хромосомной патологией. Большинство из них заканчивается выкидышем. Другими факторами являются перенесенные инфекционные заболевания, хронические болезни, вредные привычки. Однако их роль в диагностике значительно более низкая. Точного ответа на вопрос, будет ли зачат ребенок с синдромом Эдвардса, этот метод тоже не дает.
- Генетический анализ родителей . Если предыдущие методы сводились к опросу родителей, то генетический анализ представляет собой полноценное исследование, которое требует наличия специальной аппаратуры, реактивов и квалифицированных специалистов. У родителей берется кровь, из которой в лаборатории выделяют лейкоциты. После обработки специальными веществами в этих клетках становятся хорошо видны хромосомы на стадии деления. Таким образом составляется кариотип родителей. В подавляющем большинстве случаев он нормальный (при хромосомных нарушениях, которые могут быть здесь обнаружены, вероятность продолжения рода ничтожно мала ). Кроме того с помощью специальных маркеров (фрагменты молекулярных цепочек ) можно обнаружить участки ДНК с дефектными генами. Однако здесь будут обнаружены не хромосомные нарушения, а генетические мутации, которые не влияют напрямую на вероятность синдрома Эдвардса. Таким образом, генетический анализ родителей до момента зачатия, несмотря на сложность и высокую стоимость, также не дает однозначного ответа относительно прогнозов на данную патологию.
Диагностика во время внутриутробного развития
В период внутриутробного развития существует несколько способов, которые могут прямо или косвенно подтвердить наличие у зародыша хромосомной патологии. Точность этих методов значительно выше, так как врачи имеют дело не с родителями, а с самим организмом плода. Для изучения доступен как сам зародыш, так и его клетки с собственным ДНК. Данный этап называется также пренатальной диагностикой и является наиболее важным. В это время можно подтвердить диагноз, предупредить родителей о наличии патологии и при необходимости прервать беременность. Если же женщина решит рожать и новорожденный будет жив, то врачи получат возможность заранее подготовиться к оказанию ему необходимой помощи.
Основными методами исследования в рамках пренатальной диагностики являются:
- Ультразвуковое исследование (УЗИ ) . Данный метод является неинвазивным, то есть не предполагает повреждения тканей матери или плода. Он полностью безопасен и рекомендован для всех беременных женщин в рамках пренатальной диагностики (независимо от их возраста или повышенного риска для хромосомных заболеваний ). Стандартная программа предполагает, что УЗИ надо делать трижды (на 10 – 14, 20 – 24 и 32 – 34 неделе беременности ). Если лечащий врач предполагает возможность врожденных аномалий развития, могут быть проведены и незапланированные УЗИ. О синдроме Эдвардса может говорить отставание плода в размерах и массе, большое количество околоплодных вод, видимые аномалии развития (микроцефалия, деформация костей ). Эти нарушения с высокой вероятностью говорят о тяжелых генетических заболеваниях, но синдром Эдвардса окончательно подтвердить все же невозможно.
- Амниоцентез . Амниоцентез представляет собой цитологический (клеточный ) анализ околоплодных вод. Врач аккуратно вводит специальную иглу под контролем аппарата УЗИ. Прокол делается в месте, где нет петель пупочного канатика. С помощью шприца берется необходимое для исследования количество амниотической жидкости. Процедуру можно проводить во всех триместрах беременности, но оптимальным сроком для диагностики хромосомных нарушений является период после 15 недели беременности. Частота осложнений (вплоть до спонтанного прерывания беременности ) составляет до 1%, поэтому процедуру не стоит проводить при отсутствии каких-либо показаний. После забора околоплодных вод проводится обработка полученного материала. В них жидкости имеются клетки с поверхности кожи малыша, которые содержат образцы его ДНК. Именно их и проверяют на наличие генетических заболеваний.
- Кордоцентез . Кордоцентез представляет собой наиболее информативный метод пренатальной диагностики. После обезболивания и под контролем аппарата УЗИ врач прокалывает с помощью специальной иглы сосуд, проходящий в пуповине. Таким образом, получают образец крови (до 5 мл ) развивающегося ребенка. Техника выполнения анализа аналогична таковой для взрослых. Данный материал можно с высокой точностью исследовать на предмет различных генетических аномалий. В том числе можно сделать кариотипирование плода. При наличии дополнительной 18-й хромосомы можно говорить о подтвержденном синдроме Эдвардса. Данный анализ рекомендуется проводить после 18-й недели беременности (оптимально 22 – 25 недели ). Частота возможных осложнений после кордоцентеза составляет 1,5 – 2%.
- Биопсия хориона. Хорион представляет собой одну из зародышевых оболочек, содержащую клетки с генетической информацией плода. Данное исследование предполагает пункцию матки под наркозом через переднюю брюшную стенку. С помощью специальных биопсийных щипцов берут образец ткани для анализа. Затем проводится стандартное генетическое исследование полученного материала. Для диагностики синдрома Эдвардса делается кариотипирование. Оптимальным сроком для проведения биопсии хориона считают 9 – 12 неделю беременности. Частота осложнений составляет 2 – 3%. Основным преимуществом, отличающим его от других методов, является скорость получения результата (уже через 2 – 4 дня ).
Диагностика после рождения
Диагностика синдрома Эдвардса после рождения является наиболее легкой, быстрой и точной. К сожалению, на этот момент уже произошло появление на свет ребенка с тяжелой генетической патологией, эффективного лечения которой в наше время пока не существует. Если на этапе пренатальной диагностики заболевание не было выявлено (либо соответствующие исследования не проводились ), то подозрение на синдром Эдвардса появляется сразу после рождения. Ребенок обычно доношен или даже переношен, но его масса все равно ниже среднего показателя. Кроме того, обращают на себя внимание некоторые врожденные дефекты, о которых говорилось выше. Если их замечают, проводят генетический анализ для подтверждения диагноза. У ребенка берется кровь для анализа. Однако на данном этапе подтвердить наличие синдрома Эдвардса – неглавная проблема.
Основной задачей при рождении ребенка с этой патологией является обнаружение аномалий в развитии внутренних органов, которые обычно приводят к смерти в первые месяцы жизни. Именно на их поиск направлено большинство диагностических процедур непосредственно после рождения.
Для обнаружения пороков в развитии внутренних органов применяют следующие методы исследования:
- ультразвуковое исследование брюшной полости;
- эхокардиография;
- общий анализ крови и биохимический анализ крови;
- общий анализ мочи;
- компьютерная томография или магнитно-резонансная томография;
- рентгенография.
Для рентгеновского обследования младенческий возраст обычно считается противопоказанием, но в данном случае опасностью можно пренебречь. Дело в том, что речь идет об обнаружении патологий, которые представляют опасность для жизни непосредственно в данный момент. Поэтому врачи должны срочно получить всю необходимую информацию об имеющихся пороках развития. Это поможет выбрать тактику лечения. В большинстве случаев грамотное ведение пациента и соответствующий уход помогают продлить жизнь ребенка.
Таким образом, можно заключить, что существует много методов диагностики синдрома Эдвардса. Некоторые из них (амниоцентез, кордоцентез и др. ) представляют определенную опасность осложнений и не проводятся без специальных показаний. Основными показаниями считается наличие в роду случаев хромосомных заболеваний и возраст матери старше 35 лет. Программа диагностики и ведения пациентки на всех этапах беременности может быть изменена лечащим врачом по необходимости.
Прогноз для детей с синдромом Эдвардса
 Учитывая множественные нарушения развития, которые присущи синдрому Эдвардса, прогноз для новорожденных с этим диагнозом почти всегда неблагоприятный. Статистические данные (из различных независимых исследований
) говорят, что больше половины детей (50 – 55%
) не доживают до трехмесячного возраста. Первый день рождения удается отпраздновать меньше чем десяти процентам малышей. Те дети, которые доживают до старшего возраста, имеют серьезнейшие проблемы со здоровьем и нуждаются в постоянном уходе. Для продления жизни нередко необходимы сложные хирургические операции на сердце, почках или других внутренних органах. Исправление врожденных дефектов и постоянный квалифицированный уход, по сути, являются единственным лечением. У детей с классической формой синдрома Эдвардса (полной трисомией 18
) шансов на нормальное детство или сколько-нибудь длительную жизнь практически нет.
Учитывая множественные нарушения развития, которые присущи синдрому Эдвардса, прогноз для новорожденных с этим диагнозом почти всегда неблагоприятный. Статистические данные (из различных независимых исследований
) говорят, что больше половины детей (50 – 55%
) не доживают до трехмесячного возраста. Первый день рождения удается отпраздновать меньше чем десяти процентам малышей. Те дети, которые доживают до старшего возраста, имеют серьезнейшие проблемы со здоровьем и нуждаются в постоянном уходе. Для продления жизни нередко необходимы сложные хирургические операции на сердце, почках или других внутренних органах. Исправление врожденных дефектов и постоянный квалифицированный уход, по сути, являются единственным лечением. У детей с классической формой синдрома Эдвардса (полной трисомией 18
) шансов на нормальное детство или сколько-нибудь длительную жизнь практически нет.
При частичной трисомии или мозаичной форме синдрома прогноз несколько лучше. Средняя продолжительность жизни при этом увеличивается до нескольких лет. Это объясняется тем, то аномалии развития при более легких формах не ведут так быстро к смерти ребенка. Тем не менее, основная проблема, а именно серьезное отставание в умственном развитии, присуща всем без исключения больным. При достижении подросткового возраста нет шансов ни на продолжение потомства (половая зрелость обычно не наступает ), ни на возможность работы (даже механической, не требующей особых навыков ). Существуют специальные центры для ухода за детьми с врожденными заболеваниями, где больным с синдромом Эдвардса обеспечивают уход и по возможности способствуют их интеллектуальному развитию. При достаточных усилиях со стороны врачей и родителей ребенок, проживший больше года, может научиться улыбаться, реагировать на движение, самостоятельно поддерживать положение тела или питаться (при отсутствии пороков системы пищеварения ). Таким образом, признаки развития все же наблюдаются.
Высокая детская смертность при данном заболевании объясняется большим количеством пороков развития внутренних органов. Они незаметны непосредственно при рождении, но присутствуют практически у всех больных. В первые месяцы жизни дети обычно умирают от остановки сердца или дыхания.
Чаще всего пороки развития наблюдаются в следующих органах и системах:
- опорно-двигательный аппарат (кости и суставы, включая череп );
- сердечно-сосудистая система;
- центральная нервная система;
- пищеварительная система;
- мочеполовая система;
- другие нарушения.
Опорно-двигательный аппарат
Основными пороками в развитии опорно-двигательного аппарата является аномальное положение пальцев и искривление стоп. В бедренном суставе наблюдается сведение ног таким образом, что колени почти соприкасаются, а стопы смотрят немного в стороны. Нередко у детей с синдромом Эдвардса обнаруживается необычно короткая грудина. Это деформирует грудную клетку в целом и создает проблемы с дыханием, которые усугубляются по мере роста, даже если сами легкие не затронуты.
Дефекты развития черепа являются в основном косметическими. Однако такие пороки как волчья пасть, заячья губа и высокое небо создают серьезные трудности с кормлением ребенка. Нередко до проведения операций по исправлению этих дефектов ребенка переводят на парентеральное питание (в виде капельниц с питательными растворами ). Другим вариантом является использование гастростомы – специального зонда, через который пища попадает прямо в желудок. Его установление требует отдельного хирургического вмешательства.
В целом пороки развития опорно-двигательного аппарата не создают прямой угрозы для жизни ребенка. Однако косвенно они влияют на его рост и развитие. Частота таких изменений у больных синдромом Эдвардса составляет около 98%.
Сердечно-сосудистая система
Пороки развития сердечно-сосудистой системы являются основной причиной смерти в раннем детском возрасте. Дело в том, что подобные нарушения встречаются почти в 90% случаев. Чаще всего они серьезно нарушают процесс транспорта крови по организму, приводя к выраженной сердечной недостаточности. Большинство сердечных патологий может быть исправлено хирургическим путем, но не каждому ребенку можно провести такую сложную операцию.
Наиболее частыми аномалиями со стороны сердечно-сосудистой системы являются:
- незаращение межпредсердной перегородки;
- незаращение межжелудочковой перегородки;
- сращение створок клапанов (или, наоборот, их недоразвитие );
- коарктация (сужение ) аорты.
Все эти пороки сердца ведут к серьезным нарушениям кровообращения. Артериальная кровь не поступает в нужном объеме к тканям, из-за чего клетки организма начинают гибнуть.
Центральная нервная система
Пищеварительная система
Частота пороков пищеварительной системы при синдроме Эдвардса составляет до 55%. Чаще всего эти аномалии развития представляют серьезную угрозу жизни ребенка, поскольку не позволяют ему нормально усваивать питательные вещества. Питание же в обход естественных органов пищеварения сильно ослабляет организм и усугубляет состояние ребенка.
Наиболее частыми пороками развития со стороны пищеварительной системы являются:
- дивертикул Меккеля (слепой отросток в тонкой кишке );
- атрезия пищевода (зарастание его просвета, из-за чего пища не проходит в желудок );
- атрезия желчных путей (накопление желчи в пузыре ).
Все эти патологии требуют хирургического исправления. В большинстве случаев операция помогает лишь немного продлить жизнь ребенку.
Мочеполовая система
Другие нарушения
Другими возможными нарушениями развития являются грыжи (пупочная, паховая ). Могут обнаруживаться и дисковые грыжи позвоночника, которые приведут к неврологическим проблемам. Со стороны глаз иногда наблюдается микрофтальмия (маленький размер глазных яблок ).
Совокупность этих пороков развития предопределяет высокую детскую смертность. В большинстве случаев, если синдром Эдвардса диагностирован на ранних этапах беременности, врачи рекомендуют делать аборт по медицинским показаниям. Тем не менее, окончательное решение принимает сама пациентка. Несмотря на всю серьезность заболевания и плохой прогноз многие предпочитают надеяться на лучшее. Но, к сожалению, в ближайшее время серьезных сдвигов в методах диагностики и лечения синдрома Эдвардса, судя по всему, не предвидится.
- Сущность заболевания
- Причины
- Симптоматика
- Диагностика
- Лечение
- Прогнозы
После синдрома Дауна, синдром Эдвардса - самое распространённое хромосомное заболевание. Так как прогнозы для деток, страдающих такой патологией, самые неутешительные (летальный исход либо глубокая олигофрения до конца жизни), родителям обязательно нужно знать, что это за отклонение, когда его можно продиагностировать, чтобы своевременно принять верное решение, оставлять такого малыша в утробе или прервать беременность.
Сущность заболевания
Синдром трисомии 18, или синдром Эдвардса - это хромосомное заболевание, которое характеризуется целым комплексом множественных, очень тяжёлых пороков развития. Причиной является образование дополнительной 18-й хромосомы. Таким образом, кариотип больного с синдромом Эдвардса - три 18-тых хромосомы вместо нормальных двух. В 90% случаев это простая трисомия, реже - мозаичная или транслокация.
Формула, по которой читается кариотип синдрома Эдвардса, обозначается следующим образом: 47,ХХ, 18+ (это девочка) и 47,ХY, 18+ (это мальчик).
По страницам истории. Синдром Эдвардса впервые был описан Джоном Эдвардсом в 1960 году.
Причины
Основная причина заболевания - утроенная (а не сдвоенная, как в норме) 18 хромосома в кариотипе зиготы. Однако, что именно провоцирует такую патологию, генетикам до сих пор неизвестно. Тем более, что появляется лишняя хромосома ещё до оплодотворения. Существует мнение среди врачей, что никакие факты, кроме обычной случайности, здесь не виноваты. С другой стороны, существуют факторы риска, которые позволяют выделить в особую группу предполагаемые причины синдрома Эдвардса, которые нужно иметь в виду парам, собирающимся стать родителями.
- Возраст матери более 45 лет. В этом случае риск синдрома Эдвардса составляет 0,7%.
- Наследственность: если в роду уже рождались детки с подобными отклонениями, очень велик риск родить малыша с патологией в кариотипе.
- Вредные привычки остаются в силе даже здесь. Некоторые учёные доказывают, что мозаичная форма синдрома Эдвардса (она наиболее лёгкая) может быть спровоцирована долгим употреблением наркотиков, огромным количеством никотина или алкоголя в организме одного из родителей.
- Длительный приём сильнодействующих лекарств.
- Инфекции половых путей.
- Радиационное облучение очень часто порождает хромосомные мутации.
Это лишь предполагаемые генетиками причины возникновения трёх 18-тых хромосом вместо двух. За незыблемый постулат принимать их не стоит. Соответственно с ними, факторы риска синдрома Эдвардса - это возраст матери, уже имеющиеся в роду хромосомные заболевания, вредные привычки.
По сравнению с тем же синдромом Дауна, эта патология - достаточно редкое явление. Многие родители задаются вопросом, как часто ставят синдром Эдвардса: согласно статистике, 1 больной ребёнок рождается на 7 000 здоровых малышей. Так как прогнозы для таких деток совсем неутешительны, о диагнозе лучше узнать заранее.
Любопытный факт. Пока необъяснимо, но девочек с синдромом Эдвардса рождается в 3 раза больше, чем мальчиков.
Симптоматика
Чтобы принять во время беременности правильное решение, оставить малыша с таким заболеванием или нет, родители должны чётко представлять, что собой являют дети с синдромом Эдвардса, когда родятся. Клиническая картина патологии безрадостна и тяжела для сердца матери и отца. Основные симптомы можно заметить уже с момента рождения малыша.
При рождении:
- низкий вес (около 2 кг) и пренатальная гипотрофия при нормальной и даже превышенной длительности беременности;
- асфиксия.
Деформации лица и черепа:
- аномалии черепа, который имеет долихоцефалическую форму, низкий лоб, выступающий затылок;
- очень маленькие ротовое отверстие и нижняя челюсть;
- расщелины нёба и верхней губы;
- косоглазие;
- деформация ушных раковин, их низкое расположение относительно лица, вытянутость в горизонтальной плоскости;
- отсутствие мочки и козелка;
- суженный наружный слуховой проход, в ряде случаев он вообще отсутствует;
- птоз - обвисание кожи;
- короткая шея с характерной воротниковой кожной складкой.
Деформации скелета:
- короткая грудина, из-за чего уменьшены межреберные промежутки, а грудная клетка короче и шире нормальной;
- в 80 % случаев - аномальное развитие стоп: пятки резко выступают, своды провисают (стопы-качалки), большие пальцы ног утолщены и укорочены;
- скрещённые пальцы рук;
- врождённый вывих бедра;
- косолапость.
Патологии внутренних органов:
- пороки сердца и сосудов;
- гипоплазия мозжечка;
- патологии ЖКТ;
- проблемы с мочеполовой системой;
- нарушения в работе ЦНС;
- ярко выраженная умственная отсталость.
Общее развитие:
- трудности с глотанием, сосанием, дыханием.
Ребёнок с синдромом Эдвардса никогда не сможет жить полноценной жизнью, как все остальные. Всю свою короткую жизнь он будет преодолевать множество препятствий, причём совершенно не будет осознавать своей болезни. Труднее всего приходится родителям, которые вынуждены ежедневно наблюдать за развитием патологий и пороков, с которыми появился на свет их кроха. Когда же проводится диагностика данного заболевания?
Вот это да! Генетик Эдвардс, описавший данное заболевание, выделил свыше 130 симптоматических дефектов, характерных для этого синдрома.
Диагностика
Очень большое значение имеет диагностика синдрома Эдвардса, так как данная хромосомная патология - медицинское показание для прерывания беременности. Для выявления заболевания используются следующие методики.
УЗИ и допплерография
Существуют особые признаки синдрома Эдвардса на УЗИ, которые должен заметить врач:
- множественные аномалии развития плода;
- агенезия пупочной артерии;
- малая величина плаценты;
- многоводие.
Но всё это косвенные признаки синдрома Эдвардса при беременности, которые должны быть подтверждены данными других исследований.
Стандартный пренатальный скрининг
Предполагает анализ крови на наличие сывороточных маркеров:
- с 11 по 13 недели: βХГЧ и PAPP;
- с 20 по 24 недели: βХГЧ, свободный эстриола, α-фетопротеин.
Если на основании этих анализов беременная попадает в группу высокого риска, ей предлагается проведение дополнительной диагностики.
Инвазивные методы диагностики
Проводятся в исключительных случаях, когда риск рождения малыша с синдромом Эдвардса очень велик. К инвазивной диагностике относятся:
- биопсия хориона;
- амниоцентез;
- кордоцентез;
- последующее кариотипирование плода.
Если заболевание не было вовремя выявлено, и ребёнок с синдромом Эдвардса всё-таки рождается, он подвергается немедленному всестороннему обследованию.
Диагностика после рождения
Обследование больного ребёнка направлено на выявление пороков развития. Новорождённого с синдромом Эдвардса осматривают разные специалисты:
- неонатолог;
- кардиолог;
- невролог;
- хирург;
- ортопед;
- уролог и др.
Самые важные диагностические исследования, которые выполняются ребёнку с синдромом Эдвардса сразу после его рождения:
- эхокардиография;
- УЗИ почек;
- УЗИ органов брюшной полости.
Какими-то особенностями течение беременности при вынашивании ребёнка с синдромом Эдвардса не отличается. Он так же начинает шевелиться в положенные сроки, бывает даже очень активен, никаких дополнительных токсикозов не наблюдается. Поэтому выявить заболевание в этот период может только современная диагностическая аппаратура. Если же малыш всё-таки родился, вся его недолгая жизнь уйдёт на постоянное лечение сопутствующих пороков.
Мнение врачей. Девочек с синдромом Эдвардса рождается больше не потому, что они чаще всего подвержены такой патологии по 18 хромосоме. Причина - в их выживаемости. Считается, что большая часть беременностей мальчиками с таким синдромом заканчивается самопроизвольным абортом или внутриутробной гибелью плода.
Лечение
У таких деток аномалии развития и всевозможные пороки несовместимы с жизнью, поэтому лечение синдрома Эдвардса сводится к симптоматической помощи. Она направлена на поддержание элементарных физиологических функций, улучшение качества жизни и её продление, насколько это возможно. Хирургическая коррекция врождённых патологий, по мнению большинства врачей, является неоправданной и очень рискованной. В первые дни жизни лечение ограничивается такими мероприятиями, как:
- зондовое питание из-за трудностей глотания и сосания;
- длительная ИВЛ, так как имеются проблемы с дыханием.
После выписки (если это возможно) родители должны обеспечить больному малышу:
- организованный, почти профессиональный уход: хорошо, если рядом с ребёнком будет находиться человек (няня) с медицинским образованием;
- полноценное питание;
- регулярное наблюдение со стороны самых разных детских врачей.
Нужно сразу оговориться, что уход за ребёнком с синдромом Эдвардса требует очень большого терпения и сил.
Прогнозы
Родители, ребёнок которых - носитель синдрома Эдвардса, должны знать, какие прогнозы их ожидают. Как уже было сказано выше, они весьма неутешительны:
- продолжительность жизни таких деток очень невелика;
- 60% умирают до 3 месяцев (чаще всего в эту группу попадают мальчики);
- ещё 10% доживают до года;
- до 10 лет доживает только 1% родившихся;
- самая частая причина смерти - нарушения в работе сердца или остановка дыхания;
- все дети с синдромом Эдвардса до конца дней остаются глубокими олигофренами;
- организм таких деток очень ослаблен, подвержен всевозможным болезням, из-за чего они часто болеют инфекциями мочевыводящих путей, конъюнктивитом, средним отитом, синуситами, пневмониями;
- словесные навыки общения у таких детей очень ограничены;
- они могут реагировать на слова своих родителей;
- некоторые даже улыбаются, распознают и взаимодействуют с воспитателями;
- в состоянии приобрести такие навыки, как самостоятельное кормление и поднятие головки.
Если в период беременности ребёнку был поставлен диагноз синдром Эдвардса, это заставляет родителей принять ответственное решение (сроки диагностики различных патологий плода можно найти в следующей статье). Смогут ли они выхаживать тяжело больного, страдающего множественными патологиями ребёнка? Или есть смысл прервать такую беременность? Здесь только сама пара совместными усилиями может найти единственно правильный для них выход.
Во время беременности будущие мамы делают пренатальный скрининг, чтобы заблаговременно выявить генетические аномалии плода. Одним из самых тяжелых заболеваний данной группы является синдром, описанный Джоном Эдвардсом в 1960 году. В медицине оно известно как трисомия 18.
Синдром Эдвардса – что это такое простыми словами?
У здоровой мужской и женской половой клетки имеется стандартный или гаплоидный набор хромосом в количестве 23 штук. После слияния они формируют индивидуальный комплект – кариотип. Он как своеобразный ДНК-паспорт, содержит уникальные генетические данные о ребенке. Нормальный или диплоидный кариотип содержит 46 хромосом, по 2 каждого вида, от матери и отца.
При рассматриваемом заболевании в 18 паре имеется лишний дублированный элемент. Это и есть трисомия или синдром Эдвардса – кариотип, состоящий из 47 хромосом вместо 46 штук. Иногда третья копия 18 хромосомы присутствует частично или встречается не во всех клетках. Такие случаи диагностируются редко (около 5%), на течение патологии данные нюансы не влияют.
Синдром Эдвардса – причины возникновения
Генетики пока не выяснили, почему у некоторых детей происходит описываемая хромосомная мутация. Считается, что она случайна, и никаких профилактических мероприятий по ее предотвращению не разработано. Некоторые специалисты связывают внешние факторы и синдром Эдвардса – причины, предположительно способствующие развитию аномалии:
- длительное употребление наркотиков, алкоголя, табака;
- наследственность;
- возраст матери или отца старше 45 лет;
- инфекции половых органов;
- долгое применение медикаментов, воздействующих на иммунную, эндокринную и репродуктивную системы;
- подверженность радиоактивному излучению.
Синдром Эдвардса – генетика
По результатам недавних исследований в 18 хромосоме содержится 557 участков ДНК. Они кодируют более 289 видов белков в организме. В процентном соотношении это 2,5-2,6% генетического материала, поэтому так сильно влияет на развитие плода третья 18 хромосома – синдром Эдвардса повреждает кости черепа, сердечно-сосудистую и мочеполовую системы. Мутация затрагивает некоторые отделы мозга и периферические нервные сплетения. Для больного с синдромом Эдвардса характерен кариотип, представленный на рисунке. Он отчетливо показывает, что все наборы парные, кроме 18 комплекта.
Частота синдрома Эдвардса
Указанная патология встречается редко, особенно по сравнению с более известными генетическими аномалиями. Болезнь синдром Эдвардса диагностируется у одного новорожденного из 7000 здоровых младенцев, преимущественно у девочек. Нельзя утверждать, что возраст отца или материи существенно влияет на вероятность возникновения трисомии 18. Синдром Эдвардса встречается у детей всего на 0,7% чаще, если родители старше 45 лет. Эта хромосомная мутация обнаруживается и среди малышей, зачатых в молодом возрасте.
Синдром Эдвардса – признаки
Рассматриваемое заболевание имеет специфическую клиническую картину, позволяющую безошибочно определить трисомию 18. Существует две группы признаков, сопровождающих синдром Эдвардса – симптомы условно классифицируются на внутренние нарушения функционирования органов и внешние отклонения. Первый вид проявлений включает:
- пупочные, паховые грыжи;
- врожденные пороки сердца;
- отсутствие сосательного и глотательного рефлекса;
- дивертикул Меккеля;
- гастроэзофагеальный рефлюкс;
- атрезия заднего прохода или пищевода;
- гипертрофия клитора;
- недоразвитость мозолистого тела, мозжечка;
- крипторхизм;
- неправильное расположение кишечника;
- гипоспадия;
- удвоение мочеточников;
- атрофия либо сглаживание мозговых извилин;
- сегментированная или подковообразная почка;
- несформированные яичники;
- искривленный позвоночник;
- дистрофия мышц;
- низкая масса тела (около 2 кг при рождении).
Внешне тоже легко выявить синдром Эдвардса – фото малышей с трисомией 18 показывает наличие следующих признаков:
- непропорциональная маленькая голова;
- искаженная форма лица;
- узкие и короткие глазные щели;
- деформированные, низко расположенные ушные раковины (вытянуты горизонтально);
- отсутствие мочки, иногда – козелка и даже слухового прохода;
- короткая и широкая грудная клетка;
- недоразвитая нижняя челюсть;
- маленький рот, часто имеет треугольное отверстие из-за патологически укороченной верхней губы;
- вдавленная расширенная переносица;
- «стопа-качалка»;
- перепонки между пальцами или их срастание (ластообразные конечности);
- высокое нёбо, иногда – с расщелиной;
- короткая шея с выделяющейся воротниковой складкой;
- поперечные борозды и гребешки на ладонях;
- гемангиомы и папилломы на коже;
- птоз век;
- косоглазие;
- выступающий затылок и низкий лоб.
Синдром Эдвардса – диагностика
Описываемая генетическая болезнь является прямым показанием для прерывания беременности. Дети с синдромом Эдвардса никогда не смогут жить полноценно, а их здоровье будет стремительно ухудшаться. По этой причине важно диагностировать трисомию 18 на максимально раннем сроке. Для определения данной патологии разработано несколько информативных скринингов.
Анализ на синдром Эдвардса
Существуют неинвазивные и инвазивные методики исследования биологического материала. Второй тип тестов считается самым достоверным и надежным, он помогает выявить синдром Эдвардса у плода на ранних этапах развития. Неинвазивным является стандартный пренатальный скрининг крови матери. К инвазивным способам диагностики относятся:
- Биопсия ворсин хориона. Исследование проводится с 8 недели. Для осуществления анализа отщипывается кусочек оболочки плаценты, потому что его структура почти идеально совпадает с тканью плода.
- Амниоцентез. В ходе тестирования берется образец околоплодных вод. Эта процедура определяет синдром Эдвардса с 14 недели вынашивания.
- Кордоцентез. Для анализа требуется немного пуповинной крови плода, поэтому такой метод диагностики применяется исключительно на поздних сроках, с 20 недели.
Риск синдрома Эдвардса по биохимии
Пренатальный скрининг осуществляется в первом триместре беременности. Будущая мать должна сдать кровь в период с 11 по 13 неделю вынашивания для биохимического анализа. По результатам определения уровня хорионического гонадотропина и плазменного протеина А рассчитывается риск синдрома Эдвардса у плода. Если он высок, женщина вносится в соответствующую группу для следующего этапа исследований (инвазивных).
Синдром Эдвардса – признаки по УЗИ
Указанный вид диагностики используется редко, преимущественно в случаях, когда беременная не прошла предварительный генетический скрининг. Синдром Эдвардса на УЗИ можно выявить только на поздних сроках, когда плод уже почти полностью сформирован. Характерные симптомы трисомии 18:
- внутриутробные пороки сердечно-сосудистой и мочеполовой системы;
- аномалии опорно-двигательных структур;
- патологии костей черепа и мягких тканей головы.
- Косвенные признаки болезни на УЗИ:
- брадикардия;
- задержка развития плода;
- одна артерия в пуповине (должно быть две);
- грыжа в брюшной полости;
- визуальное отсутствие костей носа.
Синдром Эдвардса – лечение
Терапия рассмотренной мутации направлена на смягчение ее симптомов и облегчение жизни младенца. Излечить синдром Эдвардса и обеспечить ребенку полноценное развитие нельзя. Стандартные медицинские мероприятия помогают:
- восстановить прохождение пищи при атрезии анального отверстия или кишечника;
- организовать кормление через зонд на фоне отсутствия сосательного и глотательного рефлексов;
- стабилизировать функционирование сердечно-сосудистой системы;
- нормализовать отток мочи.
Часто синдром Эдвардса новорожденных дополнительно требует применения противовоспалительных, антибактериальных, гормональных и других сильнодействующих препаратов. Это необходимо для своевременной интенсивной терапии всех сопутствующих заболеваний, которые он провоцирует:
- опухоль Вильмса;
- конъюнктивит;
- пневмония;
- средний отит;
- легочная гипертензия;
- синусит;
- мочеполовые инфекции;
- фронтит;
- высокое артериальное давление и другое.
Синдром Эдвардса – прогноз
Большинство зародышей с описываемой генетической аномалией гибнет еще во время вынашивания из-за отторжения организмом неполноценного плода. После рождения прогноз тоже неутешителен. Если диагностирован синдром Эдвардса, сколько живут такие дети, рассмотрим в процентных соотношениях:
- 60% – не более 3-х месяцев;
- 7-10% – 1 год;
- около 1% – до 10 лет.
В исключительных случаях (частичная или мозаичная трисомия 18) единицы могут достичь зрелости. Даже в таких ситуациях синдром Джона Эдвардса будет неумолимо прогрессировать. Повзрослевшие дети с данной патологией навсегда остаются олигофренами. Максимум, чему их можно научить:
- поднимать голову;
- улыбаться;
- самостоятельно есть;
- узнавать ограниченный круг людей.
Синдром Эдвардса
трисомия 18
представляет собой тяжелое врожденное заболевание, вызванное хромосомными нарушениями. Оно является одной из наиболее распространенных патологий в данной категории (
уступает по частоте лишь синдрому Дауна
). Заболевание характеризуется многочисленными нарушениями в развитии различных органов и систем. Прогноз для ребенка обычно неблагоприятный, но многое зависит от ухода, который ему способны обеспечить родители.
Распространенность синдрома Эдвардса по земному шару варьирует от 0,015 до 0,02%. Четкой зависимости от местности или расы не наблюдается. Статистически девочки болеют в 3 – 4 раза чаще мальчиков. Научного объяснения этой пропорции пока не выявлено. Тем не менее, отмечен ряд факторов, которые могут повысить риск возникновения этой патологии.
Как и другие хромосомные мутации, синдром Эдвардса является, в принципе, неизлечимым заболеванием. Самые современные методы лечения и ухода могут лишь поддерживать жизнь ребенка и способствовать определенному прогрессу в его развитии. Единых рекомендаций по уходу за такими детьми нет из-за огромного разнообразия возможных нарушений и осложнений.
Интересные факты
- Описание основных симптомов данной болезни было сделано еще в начале XX века.
- До середины 1900-х годов собрать достаточную информацию об этой патологи не представлялось возможным. Во-первых, для этого необходим был соответствующий уровень развития технологий, который позволил бы обнаружить лишнюю хромосому. Во-вторых, большинство детей умирало в первые дни или недели жизни из-за низкого уровня оказания медицинской помощи.
- Первое полное описание болезни и ее основной причины (появление лишней 18-й хромосомы) было сделано только в 1960 году врачом Джоном Эдвардом, в честь которого тогда и назвали новую патологию.
- Реальная частота синдрома Эдвардса составляет 1 случай на 2,5 – 3 тысячи зачатий (0,03 – 0,04%), однако официальные данные значительно ниже. Это объясняется тем, что почти половина эмбрионов с данной аномалией не выживают и беременность заканчивается спонтанным абортом или внутриутробной смертью плода. Подробная диагностика причины выкидыша при этом проводится редко.
- Трисомия представляет собой вариант хромосомной мутации, при которой у человека в клетках содержится не 46, а 47 хромосом. Существует всего 3 синдрома в данной группе заболеваний. Помимо синдрома Эдвардса это синдромы Дауна (трисомия 21 хромосомы) и Патау (трисомия 13 хромосомы). При наличии других добавочных хромосом патология несовместима с жизнью. Лишь в этих трех случаях возможно рождение живого ребенка и его дальнейший (хоть и замедленный) рост и развитие.
Причины генетической патологии Синдром Эдвардса является генетическим заболеванием , которое характеризуется наличием дополнительной хромосомы в геноме человека. Чтобы разобраться в причинах, которые вызывают видимые проявления этой патологии, необходимо выяснить, что представляют собой собственно хромосомы и генетический материал в целом.
В каждой клетке человека имеется ядро, которое отвечает за хранение и обработку генетической информации. В ядре содержится 46 хромосом (
), которые представляют собой многократно упакованную молекулу
ДНКдезоксирибонуклеиновая кислота
). Эта молекула содержит в себе определенные участки, называемые генами. Каждый ген является прототипом определенного
в организме человека. При необходимости клетка считывает информацию с этого прототипа и вырабатывает соответствующий белок. Дефекты генов ведут к производству аномальных белков, которые и ответственны за появление генетических заболеваний.
Хромосомная пара состоит из двух идентичных молекул ДНК (
одна отцовская, другая – материнская
), которые сцеплены между собой небольшим мостиком (
центромерой
). Место сцепления двух хромосом в паре предопределяет форму всего соединения и его вид под микроскопом.
Все хромосомы хранят различную генетическую информацию (о разных белках) и подразделяются на следующие группы:
- группа А включает 1 – 3 пару хромосом, которые отличаются большими размерами и X-образной формой;
- группа В включает 4 – 5 пару хромосом, которые также являются крупными, но центромера лежит дальше от центра, из-за чего форма напоминает букву Х со смещенным вниз или вверх центром;
- группа С включает 6 – 12 пару хромосом, которые по форме напоминают хромосомы группы В, но уступают им по размерам;
- группа D включает 13 – 15 пару хромосом, для которых характерны средние размеры и расположение центромеры у самого конца молекул, что придает сходство с буквой V;
- группа Е включает 16 – 18 пару хромосом, которые характеризуются маленькими размерами и срединным расположением центромеры (форма буквы Х);
- группа F включает 19 – 20 хромосомные пары, которые несколько мельче хромосом группы Е и схожи с ними по форме;
- группа G включает 21 – 22 пару хромосом, которые характеризуются V-образной формой и очень маленькими размерами.
Вышеперечисленные 22 пары хромосом называются соматическими или аутосомами. Кроме того существуют половые хромосомы, которые и составляют 23 пару. Они не похожи по внешнему виду, поэтому каждая из них обозначается отдельно. Женская половая хромосома обозначается Х и сходна с группой С. Мужская половая хромосома обозначается Y и сходна по форме и размерам с группой G. Если у ребенка обе хромосомы женские (тип ХХ), то рождается девочка. Если же одна из половых хромосом женская, а другая мужская, то рождается мальчик (тип ХY). Хромосомная формула называется кариотипом и может быть обозначена следующим образом - 46,ХХ. Здесь число 46 обозначает общее количество хромосом (23 пары), а ХХ – формулу половых хромосом, которая зависит от пола (в примере представлен кариотип нормальной женщины).
Синдром Эдвардса относится к так называемым хромосомным заболеваниям, когда проблема состоит не в дефекте гена, а в дефекте целой молекулы ДНК. Если быть более точным, то классическая форма этого заболевания подразумевает наличие лишней 18-й хромосомы. Кариотип в таких случаях обозначается как 47,ХХ, 18+ (
для девочки
) и 47,ХY, 18+ (
для мальчика
). Последняя цифра обозначает номер добавочной хромосомы. Излишек генетической информации в клетках приводит к появлению соответствующих проявлений болезни, которые и объединены под названием «синдром Эдвардса». Наличие дополнительной (
) хромосомы под номером 18 дало другое (
более научное
) название болезни – трисомия 18.
В зависимости от формы хромосомного дефекта различают три вида данного заболевания:
- Полная трисомия 18 . Полная или классическая форма синдрома Эдвардса предполагает, что все клетки в организме имеют дополнительную хромосому. Данный вариант заболевания встречается более чем в 90% случаев и является наиболее тяжелым.
- Частичная трисомия 18 . Частичная трисомия 18 является весьма редким феноменом (не более 3% от всех случаев синдрома Эдвардса). При ней в клетках организма содержится не целая дополнительная хромосома, а лишь ее фрагмент. Такой дефект может быть результатом неправильного деления генетического материала, но встречается он очень редко. Иногда часть восемнадцатой хромосомы присоединяется к другой молекуле ДНК (внедряется в ее структуру, удлиняя молекулу, или просто «цепляется» с помощью мостика). Последующее деление клеток приводит к тому, что в организме имеется 2 нормальные хромосомы номер 18 и еще часть генов с этих хромосом (сохранившийся фрагмент молекулы ДНК). В этом случае количество врожденных дефектов будет намного ниже. Наблюдается избыток не всей генетической информации, закодированной в 18-й хромосоме, а лишь ее части. Для пациентов с частичной трисомией 18 прогноз лучше, чем для детей с полной формой, но все равно остается неблагоприятным.
- Мозаичная форма . Мозаичная форма синдрома Эдвардса встречается в 5 – 7% случаев данного заболевания. Механизм ее появления отличается от других видов. Дело в том, что здесь дефект образовался уже после слияния сперматозоида и яйцеклетки. Обе гаметы (половые клетки) изначально имели нормальный кариотип и несли по одной хромосоме каждого вида. После слияния сформировалась клетка с нормальной формулой 46,ХХ или 46,XY. В процессе деления этой клетки произошел сбой. При удвоении генетического материала один из фрагментов получил дополнительную 18-ю хромосому. Таким образом, на определенном этапе сформировался зародыш, часть клеток которого имеют нормальный кариотип (например, 46,ХХ), а часть – кариотип синдрома Эдвардса (47,ХХ, 18+). Доля патологических клеток никогда не превышает 50%. Их число зависит от того, на каком этапе деления начальной клетки произошел сбой. Чем позже это происходит, тем меньше будет доля дефектных клеток. Форма получила название из-за того, что все клетки организма представляют собой своеобразную мозаику. Часть из них здорова, а часть – с тяжелой генетической патологией. Закономерности в распределении клеток в организме при этом не наблюдается, то есть все дефектные клетки не могут локализоваться только в одном месте, чтобы их можно было удалить. Общее состояние пациента при этом легче, чем при классической форме трисомии 18.
Наличие дополнительной хромосомы в геноме человека представляет множество проблем. Дело в том, что клетки человека запрограммированы считывать генетическую информацию и дублировать лишь заданное природой количество молекул ДНК. Нарушения даже в структуре одного гена могут привести к серьезным заболеваниям. При наличии же целой молекулы ДНК развиваются множественные нарушения еще на этапе внутриутробного развития до рождения ребенка.
Согласно последним исследованиям хромосома номер 18 содержит 557 генов, которые кодируют не менее 289 различных белков. В процентном отношении это примерно 2,5% всего генетического материала. Нарушения, которые вызывает столь большой дисбаланс, очень серьезны. Неправильное количество белков предопределяет множество аномалий в развитии различных органов и тканей. В случае синдрома Эдвардса чаще других страдают кости черепа, некоторые отделы нервной системы, сердечно-сосудистая и мочеполовая система. По всей видимости, это связано с тем, что гены, расположенные на этой хромосоме, имеют отношение к развитию именно этих органов и систем.
Таким образом, основной и единственной причиной синдрома Эдвардса является наличие дополнительной молекулы ДНК. Наиболее часто (
при классической форме болезни
) она наследуется от одного из родителей. В норме каждая гамета (
сперматозоид и яйцеклетка
) содержат по 22 непарные соматические хромосомы, плюс одна половая. Женщина всегда передает ребенку стандартный набор 22+Х, а мужчина может передать 22+Х либо 22+Y. Это предопределяет пол ребенка. Половые клетки родителей образуются в результате разделения обычных клеток на два набора. В норме материнская клетка делится на две равные части, но иногда не все хромосомы делятся пополам. Если 18-я пара не разошлась по полюсам клетки, то одна из яйцеклеток (
или одни из сперматозоидов
) будет заранее дефектным. В нем будет не 23, а 24 хромосомы. В случае если именно эта клетка будет участвовать в оплодотворении, ребенок получит дополнительную 18-ю хромосому.
На неправильное деление клеток могут повлиять следующие факторы:
- Возраст родителей . Доказано, что вероятность хромосомных аномалий увеличивается прямо пропорционально с возрастом матери. При синдроме Эдвардса эта связь менее выражена, чем при других похожих патологиях (например, синдром Дауна). Но для женщин старше 40 лет риск родить ребенка с данной патологией в среднем в 6 – 7 раз выше. Подобная зависимость от возраста отца наблюдается в значительно меньшей степени.
- Курение и алкоголь . Такие вредные привычки как курение и злоупотребление алкоголем могут действовать на половую систему человека, влияя на деление половых клеток. Таким образом, регулярное употребление этих веществ (а также других наркотических препаратов) повышает риск неправильного распределения генетического материала.
- Прием лекарственных средств . Некоторые лекарственные препараты при неправильном приеме в первом триместре могут повлиять на деление зародышевых клеток и спровоцировать мозаичную форму синдрома Эдвардса.
- Заболевания половой сферы. Перенесенные инфекции с поражением репродуктивных органов могут отразиться на правильном делении клеток. Они повышают риск хромосомных и генетических заболеваний в целом, хотя специально для синдрома Эдвардса подобные исследования не проводились.
- Радиационное излучение. Облучение половых органов рентгеновским излучением или другими ионизирующими излучениями может повлечь генетические мутации. Особенно опасно такое внешнее воздействие в подростковом возрасте, когда деление клеток происходит наиболее активно. Частицы, формирующие излучение, легко проникают сквозь ткани и подвергают молекулу ДНК своеобразной «бомбардировке». Если это происходит в момент деления клетки, риск хромосомной мутации особенно высок.
В целом же нельзя сказать, что причины развития синдрома Эдвардса окончательно известны и хорошо изучены. Вышеперечисленные факторы лишь повышают риск развития данной мутации. Не исключается и врожденная предрасположенность некоторых людей к неправильному распределению генетического материала в половых клетках. Например, считается, что у супружеской пары, которая уже родила ребенка с синдромом Эдвардса, вероятность появления второго ребенка с аналогичной патологией составляет аж 2 – 3% (примерно в 200 раз выше, чем средняя распространенность этой болезни).
Как выглядят новорожденные с синдромом Эдвардса?
Как известно, диагностировать синдром Эдвардса можно до рождения, но в большинстве случаев данное заболевание обнаруживают непосредственно после рождения ребенка. У новорожденных с данной патологией имеется ряд ярко выраженных аномалий развития, которые иногда позволяют сразу заподозрить правильный диагноз. Подтверждение проводится позже при помощи специального генетического анализа.
Новорожденные с синдромом Эдвардса имеют следующие характерные аномалии развития:
- изменение формы черепа;
- изменение формы ушных раковин;
- аномалии развития неба;
- стопа-качалка;
- аномальная длина пальцев;
- изменение формы нижней челюсти;
- сращение пальцев;
- аномалии развития половых органов;
- флексорное положение кистей;
- дерматоглифические признаки.
Изменение формы черепа Типичным симптомом при синдроме Эдвардса является долихоцефалия. Так называется характерное изменение формы головы новорожденного ребенка, которое встречается и при некоторых других генетических заболеваниях. У долихоцефалов (детей с данным симптомом) более длинный и узкий череп. Точно подтверждается наличие этой аномалии с помощью специальных измерений. Определяют соотношение ширины черепа на уровне теменных костей к длине черепа (от выступа над переносицей до затылочного бугра). Если полученное соотношение меньше 75%, то данный ребенок относится к долихоцефалам. Сам по себе данный симптом не является серьезным нарушением. Это просто один из видов формы черепа, встречающийся и у абсолютно нормальных людей. Дети с синдромом Эдвардса в 80 – 85% случаев являются выраженными долихоцефалами, у которых диспропорцию длины и ширины черепа можно заметить и без специальных измерений.
Другим вариантом аномалии развития черепа является так называемая микроцефалия, при которой размеры головы в целом слишком малы по сравнению с остальным туловищем. Прежде всего, это касается не лицевого черепа (
челюсти, скулы, глазницы
), а именно черепной коробки, в которой располагается мозг. Микроцефалия менее характерна для синдрома Эдвардса, чем долихоцефалия, но она тоже встречается с более высокой частотой, чем среди здоровых людей.
Изменение формы ушной раковины
Если долихоцефалия может быть вариантом нормы, то патологии развития ушной раковины у детей с синдромом Эдвардса куда тяжелее. В определенной степени этот симптом наблюдается более чем у 95% детей с полной формой данного заболевания. При мозаичной форме его частота несколько меньше. Ушная раковина обычно располагается ниже, чем у нормальных людей (
иногда ниже уровня глаз
). Характерные выпуклости хряща, который формирует ушную раковину, плохо выражены или отсутствуют. Также могут отсутствовать мочка или козелок (
небольшой выступающий участок хряща спереди от слухового отверстия
). Сам слуховой проход обычно сужен, а примерно в 20 – 25% - вовсе отсутствует.
Аномалии развития неба Небные отростки верхней челюсти в процессе развития эмбриона срастаются, формируя твердое небо. У детей с синдромом Эдвардса этот процесс нередко остается незавершенным. В том месте, где у нормальных людей располагается срединный шов (его можно прощупать посередине твердого неба языком) у них идет продольная щель.
Существует несколько вариантов данного дефекта:
- незаращение мягкого неба (задняя, глубокая часть неба, которая нависает над глоткой);
- частичное незаращение твердого неба (щель не тянется на протяжении всей верхней челюсти);
- полное незаращение твердого и мягкого неба;
- полное незаращение неба и губы.
В ряде случаев расщепление неба является двусторонним. Два выступающих вверх уголка верхней губы являются началом патологических щелей. Ребенок не может полностью закрыть рот из-за этого дефекта. В тяжелых случаях явно видно сообщение ротовой и носовой полости (даже при закрытом рте). Передние зубы в будущем могут отсутствовать или расти в сторону.
Данные дефекты развития известны также под названием волчья пасть, расщепление неба, заячья губа. Все они могут встречаться и не в рамках синдрома Эдвардса, однако у детей с этой патологией их частота особенно высока (
почти 20% новорожденных
). Значительно чаще (
до 65% новорожденных
) обладают другой особенностью, известной как высокое или готическое небо. Оно может быть отнесено к вариантам нормы, так как встречается и у здоровых людей.
Наличие расщепленного верхнего неба или верхней губы еще не подтверждает синдром Эдвардса. Этот порок развития может встречаться с довольно высокой частотой и самостоятельно без сопутствующих нарушений со стороны других органов и систем. Для исправления данной аномалии существует ряд стандартных хирургических вмешательств.
Стопа-качалка
Так называется характерное изменение стопы, которое встречается, в основном, в рамках синдрома Эдвардса. Частота его при данном заболевании достигает 75%. Дефект заключается в неправильном взаимоположении таранной, пяточной и ладьевидной костей. Его относят к категории плоско-вальгусных деформаций стопы у детей.
Внешне стопа у новорожденного ребенка выглядит следующим образом. Пяточный бугор, на который опирается задняя часть стопы, выдается назад. Свод при этом может полностью отсутствовать. Это легко заметить, посмотрев на стопу с внутренней стороны. В норме там вырисовывается вогнутая линия, направляющаяся от пятки к основанию большого пальца. При стопе-качалке этой линии нет. Стопа плоская или даже выпуклая. Это и придает ей сходство с ножками кресла-качалки.
Аномальная длина пальцев
У детей с синдромом Эдвардса на фоне изменений в строении стопы может наблюдаться ненормальная пропорция в длине пальцев ног. В частности, речь идет о большом пальце, который в норме является самым длинным. У новорожденных с данным синдромом он уступает по длине второму пальцу. Данный дефект можно заметить лишь при распрямлении пальцев и тщательном их рассмотрении. С возрастом, по мере роста ребенка, он становится более заметным. Поскольку укорочение большого пальца стопы встречается в основном при стопе-качалке, распространенность этих симптомов у новорожденных примерно одинакова.
У взрослых укорочение большого пальца на ноге не имеет такой диагностической ценности. Подобный дефект может быть индивидуальной особенностью у здорового человека или следствием воздействия других факторов (
деформация суставов, заболевания костей, ношение обуви, не соответствующей по размеру
). В связи с этим данный признак нужно рассматривать как возможный симптом только у новорожденных детей при наличии других аномалий развития.
Изменение формы нижней челюсти
Изменения формы нижней челюсти у новорожденных встречаются почти в 70% случаев. В норме подбородок у детей не выступает вперед так, как у взрослых, но у больных с синдромом Эдвардса он слишком уж сильно втянут. Это происходит из-за недоразвития нижней челюсти, которое носит название микрогнатия (
микрогения
). Данный симптом встречается и при других врожденных заболеваниях. Не так уж редко можно встретить и взрослых людей с похожими чертами лица. При отсутствии сопутствующих патологий это считается вариантом нормы, хоть и ведет к некоторым трудностям.
У новорожденных с микрогнатией обычно быстро появляются следующие проблемы:
- невозможность долго держать рот закрытым (подтекание слюны);
- затруднения при кормлении;
- позднее развитие зубов и неправильное их расположение.
Зазор между нижней и верхней челюстью может составлять более 1 см, что очень много, учитывая размеры головы малыша.
Сращение пальцев
Сращение пальцев, или по-научному синдактилия, наблюдается приблизительно у 45% новорожденных. Чаще всего эта аномалия затрагивает пальцы ног, но встречается и синдактилия на руках. В легких случаях сращение образовано кожной складкой наподобие короткой перепонки. В более тяжелых случаях наблюдается сращение мостиками костной ткани.
Синдактилия встречается не только при синдроме Эдвардса, но и при многих других хромосомных заболеваниях. Известны и случаи, когда этот порок развития являлся единственным, и в остальном больной ничем не отличался от нормальных детей. В связи с этим сращение пальцев является лишь одним из возможных признаков синдрома Эдвардса, который помогает заподозрить диагноз, но не подтверждает его.
Аномалии развития половых органов
Непосредственно после
у новорожденных с синдромом Эдвардса иногда можно наблюдать аномалии развития внешних половых органов. Как правило, они сочетаются с дефектами развития всего мочеполового аппарата, однако без специальных диагностических мероприятий это установить невозможно. Наиболее же частыми аномалиями, заметными внешне, являются недоразвитие полового члена у мальчиков и гипертрофия (
увеличение в размерах
) клитора у девочек. Они встречаются примерно в 15 – 20% случаев. Несколько реже может наблюдаться аномальное расположение мочеиспускательного канала (
гипоспадия
) или отсутствие яичек в мошонке у мальчиков (
крипторхизм
Флексорное положение кистей
Флексорное положение кистей – это особое расположение пальцев, вызванное не столько структурными нарушениями в области кисти, сколько повышенным тонусом мышц. Сгибатели пальцев и кисти постоянно напряжены, из-за чего большой палец и мизинец как бы прикрывают остальные пальцы, которые при этом прижаты к ладони. Данный симптом наблюдается при многих врожденных патологиях и не является характерным именно для синдрома Эдвардса. Тем не менее, при обнаружении кисти подобной формы необходимо предполагать эту патологию. При ней флексорное положение пальцев наблюдается почти у 90% новорожденных.
Дерматоглифические признаки При многих хромосомных аномалиях у новорожденных имеются характерные дерматоглифические изменения (аномальные узоры и складки на коже ладоней). При синдроме Эдвардса некоторые признаки можно обнаружить почти в 60% случаев. Они имеют значение в основном для предварительной диагностики при мозаичной или частичной форме болезни. При полной трисомии 18 к дерматоглифике не прибегают, так как для подозрения синдрома Эдвардса хватает других, более заметных аномалий развития.
Основными дерматоглифическими признаками синдрома Эдвардса являются:
- дуги на подушечках пальцев располагаются с большей частотой, нежели у здоровых людей;
- кожная складка между последней (ногтевой) и предпоследней (срединной) фалангами пальцев отсутствует;
- у 30% новорожденных на ладони имеется так называемая поперечная борозда (обезьянья линия, линия Симиан).
Специальные исследования могут выявить и другие отклонения от нормы, однако непосредственно после рождения, без привлечения узких специалистов, врачам достаточно этих изменений.
Помимо вышеперечисленных признаков существует еще целый ряд возможных аномалий развития, которые могут помочь в предварительной диагностике синдрома Эдвардса. По некоторым данными при подробном внешнем осмотре можно обнаружить до 50 внешних признаков. Сочетание наиболее частых симптомов, представленных выше, с высокой вероятностью говорит о наличии у ребенка этой тяжелой патологии. При мозаичном варианте синдрома Эдвардса множественных аномалий может и не быть, однако наличие даже одного из них является показанием к проведению специального генетического теста.
Как выглядят дети с синдромом Эдвардса? У детей с синдромом Эдвардса по мере взросления обычно обнаруживаются самые разные сопутствующие патологии. Их симптомы начинают проявляться уже через несколько недель после рождения. Эти симптомы могут оказаться первым проявлением синдрома, так как при мозаичном варианте в редких случаях болезнь может остаться незамеченной непосредственно после рождения. Тогда диагностика заболевания усложняется.
Большинство внешних проявлений синдрома, замеченных при рождении, остаются и становятся более заметными. Речь идет о форме черепа, стопе-качалке, деформации ушной раковины и т. п. Постепенно к ним начинают прибавляться и другие внешние проявления, которые невозможно было заметить сразу после рождения. В данном случае речь идет о признаках, которые могут появиться у детей в первый год жизни.
Дети с синдромом Эдвардса имеют следующие внешние особенности:
- отставание в физическом развитии;
- косолапость;
- аномальный тонус мышц;
- ненормальные эмоциональные реакции.
Отставание в физическом развитии Отставание в физическом развитии объясняется низкой массой тела ребенка при рождении (всего 2000 – 2200 г при нормальном сроке беременности). Значительную роль играет и генетический дефект, который не позволяет всем системам организма нормально и гармонично развиваться. Основные показатели, по которым оценивают рост и развитие ребенка, сильно снижены.
Заметить отставание ребенка можно по следующим антропометрическим показателям:
- рост ребенка;
- вес ребенка;
- окружность грудной клетки;
- окружность головы (данный показатель может быть в норме или даже увеличен, но на него нельзя полагаться из-за врожденной деформации черепа).
Косолапость Косолапость является следствием деформации костей и суставов стоп, а также отсутствия нормального контроля со стороны нервной системы. Дети с трудом начинают ходить (большинство не доживает до этого этапа из-за врожденных пороков развития). Внешне о наличии косолапости можно судить по деформации стоп, ненормальному положению ног в состоянии покоя.
Аномальный тонус мышц
Аномальный тонус, который при рождении вызывает флексорное положение кисти, по мере роста начинает проявляться и на других группах мышц. Чаще всего у детей с синдромом Эдвардса сила мышц снижена, они вялые и лишены нормального тонуса. В зависимости от характера повреждений центральной нервной системы некоторые группы могут иметь повышенный тонус, что проявляется спастическими сокращениями этих мышц (
например, сгибатели рук или разгибатели ног
). Внешне это проявляется отсутствием минимальной координации движений. Иногда спастические сокращения ведут к ненормальным перегибам конечностей или даже к вывихам.
Ненормальные эмоциональные реакции
Отсутствие или ненормальное проявление каких-либо эмоций является следствием аномалий в развитии некоторых отделов мозга (
чаще всего мозжечка и мозолистого тела
). Эти изменения приводят к серьезному отставанию в умственном развитии, которое наблюдается у всех без исключения детей с синдромом Эдвардса. Внешне низкий уровень развития проявляется характерным «отсутствующим» выражением лица, отсутствием эмоционального ответа на внешние раздражители. Ребенок плохо поддерживает визуальный контакт (
не следит за движущимся перед глазами пальцем и т. п.
). Отсутствие реакции на резкие звуки может быть следствием поражения как нервной системы, так и слухового аппарата. Все эти признаки обнаруживаются по мере роста ребенка в первые месяцы жизни.
Как выглядят взрослые с синдромом Эдвардса?
В подавляющем большинстве случаев дети, рожденные с синдромом Эдвардса, не доживают до взрослого возраста. При полной форме этого заболевания, когда лишняя хромосома присутствует в каждой клетке тела, 90% детей умирает в возрасте до 1 года из-за серьезных аномалий развития внутренних органов. Даже при условии хирургического исправления возможных дефектов и качественном уходе их организм более подвержен инфекционным заболеваниям. Этому способствуют и нарушения питания, которые встречаются у большинства детей. Все это объясняет высочайшую смертность при синдроме Эдвардса.
При более легкой мозаичной форме, когда лишь часть клеток в организме содержит аномальный набор хромосом, выживаемость несколько больше. Однако даже в этих случаях до взрослого возраста доживают единичные пациенты. Их внешний вид определяется врожденными аномалиями, которые присутствовали при рождении (
заячья губа, деформированная ушная раковина, и др.
). Основным же симптомом, присутствующим у всех без исключения детей, является серьезнейшее отставание в умственном развитии. Дожив до взрослого возраста, ребенок с синдромом Эдвардса является глубоким олигофреном (
IQ менее 20, что соответствует самой тяжелой степени умственной отсталости
). В целом же в медицинской литературе описываются единичные случаи, когда дети с синдромом Эдвардса доживали до совершеннолетнего возраста. Из-за этого накоплено слишком мало объективных данных, чтобы говорить о внешних признаках этого заболевания у взрослых.
Диагностика генетической патологии
В настоящее время существуют три основных этапа диагностики синдрома Эдвардса, каждый из которых включает несколько возможных методов. Поскольку данное заболевание является неизлечимым, родителям следует обратить внимание на возможности этих методов и воспользоваться ими. Большинство анализов проводится в специальных центрах пренатальной диагностики, где имеется вся необходимая техника для поиска генетических заболеваний. Однако даже консультация у врача-генетика или неонатолога может оказаться полезной.
Диагностика синдрома Эдвардса возможна на следующих этапах:
- диагностика до момента зачатия;
- диагностика во время внутриутробного развития;
- диагностика после рождения.
Диагностика до момента зачатия Диагностика до момента зачатия ребенка является идеальным вариантом, но, к сожалению, на современном этапе развития медицины ее возможности очень ограничены. Врачи могут с помощью нескольких методов предположить повышенную вероятность рождения ребенка с хромосомным заболеванием, но не более того. Дело в том, что при синдроме Эдвардса, в принципе, нарушения у родителей обнаружить нельзя. Дефектная половая клетка с 24 хромосомами является лишь одной из многих тысяч. Поэтому сказать наверняка до момента зачатия, родится ли ребенок с данным заболеванием, нельзя.
Основными методами диагностики до момента зачатия являются:
- Семейный анамнез . Семейный анамнез представляет собой подробный опрос обоих родителей об их родословной. Врача интересуют любые случаи наследственных (и особенно хромосомных) заболеваний в семье. Если хотя бы один из родителей припоминает случай трисомии (синдром Эдвардса, Дауна, Патау), это сильно повышает вероятность рождения больного ребенка. Однако риск все равно составляет не более 1%. При повторных случаях этих заболеваний у предков риск многократно возрастает. По сути, анализ сводится к консультации у неонатолога или генетика. Предварительно родители могут постараться собрать более подробную информацию о своих предках (желательно на 3 – 4 колена). Это повысит точность данного метода.
- Обнаружение факторов риска . Основным фактором риска, который объективно повышает риск хромосомных аномалий, является возраст матери. Как уже говорилось выше, у матерей после 40 лет вероятность рождения ребенка с синдромом Эдвардса многократно возрастает. По некоторым данным, после 45 лет (возраст матери) почти каждая пятая беременность сопровождается хромосомной патологией. Большинство из них заканчивается выкидышем. Другими факторами являются перенесенные инфекционные заболевания, хронические болезни, вредные привычки. Однако их роль в диагностике значительно более низкая. Точного ответа на вопрос, будет ли зачат ребенок с синдромом Эдвардса, этот метод тоже не дает.
- Генетический анализ родителей . Если предыдущие методы сводились к опросу родителей, то генетический анализ представляет собой полноценное исследование, которое требует наличия специальной аппаратуры, реактивов и квалифицированных специалистов. У родителей берется кровь, из которой в лаборатории выделяют лейкоциты. После обработки специальными веществами в этих клетках становятся хорошо видны хромосомы на стадии деления. Таким образом составляется кариотип родителей. В подавляющем большинстве случаев он нормальный (при хромосомных нарушениях, которые могут быть здесь обнаружены, вероятность продолжения рода ничтожно мала). Кроме того с помощью специальных маркеров (фрагменты молекулярных цепочек) можно обнаружить участки ДНК с дефектными генами. Однако здесь будут обнаружены не хромосомные нарушения, а генетические мутации, которые не влияют напрямую на вероятность синдрома Эдвардса. Таким образом, генетический анализ родителей до момента зачатия, несмотря на сложность и высокую стоимость, также не дает однозначного ответа относительно прогнозов на данную патологию.
Диагностика во время внутриутробного развития В период внутриутробного развития существует несколько способов, которые могут прямо или косвенно подтвердить наличие у зародыша хромосомной патологии. Точность этих методов значительно выше, так как врачи имеют дело не с родителями, а с самим организмом плода. Для изучения доступен как сам зародыш, так и его клетки с собственным ДНК. Данный этап называется также пренатальной диагностикой и является наиболее важным. В это время можно подтвердить диагноз, предупредить родителей о наличии патологии и при необходимости прервать беременность. Если же женщина решит рожать и новорожденный будет жив, то врачи получат возможность заранее подготовиться к оказанию ему необходимой помощи.
Основными методами исследования в рамках пренатальной диагностики являются:
- Ультразвуковое исследование (УЗИ) . Данный метод является неинвазивным, то есть не предполагает повреждения тканей матери или плода. Он полностью безопасен и рекомендован для всех беременных женщин в рамках пренатальной диагностики (независимо от их возраста или повышенного риска для хромосомных заболеваний). Стандартная программа предполагает, что УЗИ надо делать трижды (на 10 – 14, 20 – 24 и 32 – 34 неделе беременности). Если лечащий врач предполагает возможность врожденных аномалий развития, могут быть проведены и незапланированные УЗИ. О синдроме Эдвардса может говорить отставание плода в размерах и массе, большое количество околоплодных вод, видимые аномалии развития (микроцефалия, деформация костей). Эти нарушения с высокой вероятностью говорят о тяжелых генетических заболеваниях, но синдром Эдвардса окончательно подтвердить все же невозможно.
- Амниоцентез . Амниоцентез представляет собой цитологический (клеточный) анализ околоплодных вод. Врач аккуратно вводит специальную иглу под контролем аппарата УЗИ. Прокол делается в месте, где нет петель пупочного канатика. С помощью шприца берется необходимое для исследования количество амниотической жидкости. Процедуру можно проводить во всех триместрах беременности, но оптимальным сроком для диагностики хромосомных нарушений является период после 15 недели беременности. Частота осложнений (вплоть до спонтанного прерывания беременности) составляет до 1%, поэтому процедуру не стоит проводить при отсутствии каких-либо показаний. После забора околоплодных вод проводится обработка полученного материала. В них жидкости имеются клетки с поверхности кожи малыша, которые содержат образцы его ДНК. Именно их и проверяют на наличие генетических заболеваний.
- Кордоцентез . Кордоцентез представляет собой наиболее информативный метод пренатальной диагностики. После обезболивания и под контролем аппарата УЗИ врач прокалывает с помощью специальной иглы сосуд, проходящий в пуповине. Таким образом, получают образец крови (до 5 мл) развивающегося ребенка. Техника выполнения анализа аналогична таковой для взрослых. Данный материал можно с высокой точностью исследовать на предмет различных генетических аномалий. В том числе можно сделать кариотипирование плода. При наличии дополнительной 18-й хромосомы можно говорить о подтвержденном синдроме Эдвардса. Данный анализ рекомендуется проводить после 18-й недели беременности (оптимально 22 – 25 недели). Частота возможных осложнений после кордоцентеза составляет 1,5 – 2%.
- Биопсия хориона. Хорион представляет собой одну из зародышевых оболочек, содержащую клетки с генетической информацией плода. Данное исследование предполагает пункцию матки под наркозом через переднюю брюшную стенку. С помощью специальных биопсийных щипцов берут образец ткани для анализа. Затем проводится стандартное генетическое исследование полученного материала. Для диагностики синдрома Эдвардса делается кариотипирование. Оптимальным сроком для проведения биопсии хориона считают 9 – 12 неделю беременности. Частота осложнений составляет 2 – 3%. Основным преимуществом, отличающим его от других методов, является скорость получения результата (уже через 2 – 4 дня).
Диагностика после рождения Диагностика синдрома Эдвардса после рождения является наиболее легкой, быстрой и точной. К сожалению, на этот момент уже произошло появление на свет ребенка с тяжелой генетической патологией, эффективного лечения которой в наше время пока не существует. Если на этапе пренатальной диагностики заболевание не было выявлено (либо соответствующие исследования не проводились), то подозрение на синдром Эдвардса появляется сразу после рождения. Ребенок обычно доношен или даже переношен, но его масса все равно ниже среднего показателя. Кроме того, обращают на себя внимание некоторые врожденные дефекты, о которых говорилось выше. Если их замечают, проводят генетический анализ для подтверждения диагноза. У ребенка берется кровь для анализа. Однако на данном этапе подтвердить наличие синдрома Эдвардса – неглавная проблема.
Основной задачей при рождении ребенка с этой патологией является обнаружение аномалий в развитии внутренних органов, которые обычно приводят к смерти в первые месяцы жизни. Именно на их поиск направлено большинство диагностических процедур непосредственно после рождения.
Для обнаружения пороков в развитии внутренних органов применяют следующие методы исследования:
- ультразвуковое исследование брюшной полости;
- эхокардиография;
- общий анализ крови и биохимический анализ крови;
- общий анализ мочи;
- компьютерная томография или магнитно-резонансная томография;
- рентгенография.
Для рентгеновского обследования младенческий возраст обычно считается противопоказанием, но в данном случае опасностью можно пренебречь. Дело в том, что речь идет об обнаружении патологий, которые представляют опасность для жизни непосредственно в данный момент. Поэтому врачи должны срочно получить всю необходимую информацию об имеющихся пороках развития. Это поможет выбрать тактику лечения. В большинстве случаев грамотное ведение пациента и соответствующий уход помогают продлить жизнь ребенка.
Таким образом, можно заключить, что существует много методов диагностики синдрома Эдвардса. Некоторые из них (
амниоцентез, кордоцентез и др.
) представляют определенную опасность осложнений и не проводятся без специальных показаний. Основными показаниями считается наличие в роду случаев хромосомных заболеваний и возраст матери старше 35 лет. Программа диагностики и ведения пациентки на всех этапах беременности может быть изменена лечащим врачом по необходимости.
Прогноз для детей с синдромом Эдвардса
Учитывая множественные нарушения развития, которые присущи синдрому Эдвардса, прогноз для новорожденных с этим диагнозом почти всегда неблагоприятный. Статистические данные (
из различных независимых исследований
) говорят, что больше половины детей (
) не доживают до трехмесячного возраста. Первый день рождения удается отпраздновать меньше чем десяти процентам малышей. Те дети, которые доживают до старшего возраста, имеют серьезнейшие проблемы со здоровьем и нуждаются в постоянном уходе. Для продления жизни нередко необходимы сложные хирургические операции на
Почках или других внутренних органах. Исправление врожденных дефектов и постоянный квалифицированный уход, по сути, являются единственным лечением. У детей с классической формой синдрома Эдвардса (
полной трисомией 18
) шансов на нормальное детство или сколько-нибудь длительную жизнь практически нет.
При частичной трисомии или мозаичной форме синдрома прогноз несколько лучше. Средняя продолжительность жизни при этом увеличивается до нескольких лет. Это объясняется тем, то аномалии развития при более легких формах не ведут так быстро к смерти ребенка. Тем не менее, основная проблема, а именно серьезное отставание в умственном развитии, присуща всем без исключения больным. При достижении подросткового возраста нет шансов ни на продолжение потомства (
половая зрелость обычно не наступает
), ни на возможность работы (
даже механической, не требующей особых навыков
). Существуют специальные центры для ухода за детьми с врожденными заболеваниями, где больным с синдромом Эдвардса обеспечивают уход и по возможности способствуют их интеллектуальному развитию. При достаточных усилиях со стороны врачей и родителей ребенок, проживший больше года, может научиться улыбаться, реагировать на движение, самостоятельно поддерживать положение тела или питаться (
при отсутствии пороков системы пищеварения
). Таким образом, признаки развития все же наблюдаются.
Высокая детская смертность при данном заболевании объясняется большим количеством пороков развития внутренних органов. Они незаметны непосредственно при рождении, но присутствуют практически у всех больных. В первые месяцы жизни дети обычно умирают от остановки сердца или дыхания.
Чаще всего пороки развития наблюдаются в следующих органах и системах:
- опорно-двигательный аппарат (кости и суставы, включая череп);
- сердечно-сосудистая система;
- центральная нервная система;
- пищеварительная система;
- мочеполовая система;
- другие нарушения.
Опорно-двигательный аппарат Основными пороками в развитии опорно-двигательного аппарата является аномальное положение пальцев и искривление стоп. В бедренном суставе наблюдается сведение ног таким образом, что колени почти соприкасаются, а стопы смотрят немного в стороны. Нередко у детей с синдромом Эдвардса обнаруживается необычно короткая грудина. Это деформирует грудную клетку в целом и создает проблемы с дыханием, которые усугубляются по мере роста, даже если сами легкие не затронуты.
Дефекты развития черепа являются в основном косметическими. Однако такие пороки как волчья пасть, заячья губа и высокое небо создают серьезные трудности с кормлением ребенка. Нередко до проведения операций по исправлению этих дефектов ребенка переводят на парентеральное питание (
в виде капельниц с питательными растворами
). Другим вариантом является использование гастростомы – специального зонда, через который пища попадает прямо в желудок. Его установление требует отдельного хирургического вмешательства.
В целом пороки развития опорно-двигательного аппарата не создают прямой угрозы для жизни ребенка. Однако косвенно они влияют на его рост и развитие. Частота таких изменений у больных синдромом Эдвардса составляет около 98%.
Сердечно-сосудистая система
Пороки развития сердечно-сосудистой системы являются основной причиной смерти в раннем детском возрасте. Дело в том, что подобные нарушения встречаются почти в 90% случаев. Чаще всего они серьезно нарушают процесс транспорта крови по организму, приводя к выраженной
сердечной недостаточности
Большинство сердечных патологий может быть исправлено хирургическим путем, но не каждому ребенку можно провести такую сложную операцию.
Наиболее частыми аномалиями со стороны сердечно-сосудистой системы являются:
- незаращение межпредсердной перегородки;
- незаращение межжелудочковой перегородки;
- сращение створок клапанов (или, наоборот, их недоразвитие);
- коарктация (сужение) аорты.
Все эти пороки сердца ведут к серьезным нарушениям кровообращения. Артериальная кровь не поступает в нужном объеме к тканям, из-за чего клетки организма начинают гибнуть.
Центральная нервная система
Самым характерным пороком со стороны центральной нервной системы является недоразвитие мозолистого тела и мозжечка. Это причина самых разнообразных нарушений, в том числе и умственной отсталости, которая наблюдается у 100% детей. Кроме того, нарушения на уровне головного и спинного мозга вызывают аномальный тонус мышц и предрасположенность к
судорогам
или спастическим сокращениям мышц.
Пищеварительная система
Частота пороков пищеварительной системы при синдроме Эдвардса составляет до 55%. Чаще всего эти аномалии развития представляют серьезную угрозу жизни ребенка, поскольку не позволяют ему нормально усваивать питательные вещества. Питание же в обход естественных органов пищеварения сильно ослабляет организм и усугубляет состояние ребенка.
Наиболее частыми пороками развития со стороны пищеварительной системы являются:
- дивертикул Меккеля (слепой отросток в тонкой кишке);
- атрезия пищевода (зарастание его просвета, из-за чего пища не проходит в желудок);
- атрезия желчных путей (накопление желчи в пузыре).
Все эти патологии требуют хирургического исправления. В большинстве случаев операция помогает лишь немного продлить жизнь ребенку.
Мочеполовая система
Наиболее серьезные пороки со стороны мочеполовой системы связаны с нарушением работы почек. В ряде случаев наблюдается атрезия мочеточников. Почка с одной стороны может быть дублирована или сращена с лежащими рядом тканями. Если имеет место нарушение фильтрации, в организме со временем начинают накапливаться токсичные продукты жизнедеятельности. Кроме того может наблюдаться рост
артериального давления
и нарушения в работе сердца. Серьезные аномалии развития почек представляют прямую угрозу для жизни.
Другие нарушения
Другими возможными нарушениями развития являются
грыжи (пупочная, паховая)
Могут обнаруживаться и
дисковые грыжи позвоночника
Которые приведут к неврологическим проблемам. Со стороны глаз иногда наблюдается микрофтальмия (
маленький размер глазных яблок
Совокупность этих пороков развития предопределяет высокую детскую смертность. В большинстве случаев, если синдром Эдвардса диагностирован на ранних этапах беременности, врачи рекомендуют делать аборт по медицинским показаниям. Тем не менее, окончательное решение принимает сама пациентка. Несмотря на всю серьезность заболевания и плохой прогноз многие предпочитают надеяться на лучшее. Но, к сожалению, в ближайшее время серьезных сдвигов в методах диагностики и лечения синдрома Эдвардса, судя по всему, не предвидится.
Современные диагностические методы позволяют выявить большое количество врождённых заболеваний еще на этапе вынашивания ребенка. Синдром Эдвардса является одной из самых опасных патологий, приводящих к развитию большого количества нарушений. В медицине заболевание называют Трисомия 18.
А теперь остановимся на этом подробнее.
Что такое «синдром Эдвардса»?
Синдромом Эдвардса называют хромосомную аномалию, приводящую к возникновению целого перечня нарушений и отклонений в развитии ребенка. Изменения происходят в 18 хромосоме. У больных с синдромом наблюдаются ее лишние копии. Этот факт становится причиной появления осложнений генетической природы.
Частота возникновения заболевания составляет 1 случаев к 7000 здоровых детей. Большинство новорождённых, появившихся на свет с этим отклонением, умирают в первые недели жизни. Лишь порядка 10% детей живут один год. Болезнь приводит к возникновению глубокой умственной отсталости, врождённых поражений внутренних и внешних органов. Нередко возникают пороки сердца, почек, мозга. У детей с такой патологией маленькая голова и челюсть. Имеет место быть расщелина губы или нёба. Присутствует косолапость.
Впервые заболевание было описано в 1960 году. Симптомы болезни сформировал и описал д. Эдвардс. Он сумел установить зависимость между рядом признаков и обнаружил более 130 дефектов, сопутствующих заболеванию. Симптомы патологии проявляются весьма ярко. Однако современные методы терапии против них неэффективны. Существует три разновидности заболевания. В перечень входят:
- Простая трисомия. Заболевание выявляется в 90% случаев.
- Мозаичная трисомия. Повреждённые хромосомы присутствуют только в части клеток.
- Транслокация.
Согласно статистике, у девочек заболевания выявляют в 3 раза чаще, чем у мальчиков. Шанс появления новорождённого с синдромом Эдвардса возрастает вместе с возрастом матери. Вероятность появления такого ребенка наиболее высока, если женщине больше 30 лет. После 45 лет риск повышается до 0,7%.
Патология существенно сказывается на продолжительности жизни. Мальчики обычно доживают лишь до 3 месяцев. Больные девочки живут дольше. Отмечается, что такие младенцы достигали десятимесячного возраста. Однако к стадии взросления такие дети практически не переходит. Причинами гибели плода выступают легочные инфекции или удушья, нарушения функционирования сердца или сосудов, а также неработающей или закупоренной кишечный тракт. Все вышеуказанные явления возникают в результате патологии .
Как выглядит синдром Эдвардса с фото
Современные методы позволяют диагностировать патологию до рождения ребенка. Однако в большинстве случаев заболевание обнаруживается после появления новорождённого на свет. У детей с синдромом Эдвардса присутствуют ярко выраженные аномалии развития. Они дают возможность сразу заподозрить правильный диагноз. Подтверждение выполняется при помощи специального анализа.
Младенцев, родившихся с синдромом Эдвардса, легко отличить от других по внешнему виду. У таких детей присутствуют:
- срощенные пальцы;
- стопа-качалка
- измененная форма черепа;
- аномалии развития половых органов;
- изменение формы нижней челюсти;
- аномалия развития нёба;
- дерматоглифические признаки;
- изменение формы ушных раковин;
- флексорное положение кистей.
Типичным внешним проявлением выступает изменение формы головы новорожденного ребёнка. У детей с таким синдромом имеется более длинный и узкий череп. Аномалия подтверждается при помощи специальных измерений. Другим вариантом аномалии выступает наличие слишком маленькой головы по сравнению с остальным туловищем. Это явление затрагивает именно черепную коробку, в которой размещается мозг.
У всех детей с подобной проблемой присутствуют патологии развития ушных раковин. Этот симптом наблюдается в 95% случаев. Однако при мозаичной форме синдрома Эдвардса шанс на возникновение аномалии снижается. Ушная раковина у таких детей располагается значительно ниже. Иногда она находится ниже уровня глаз. Характерная выпуклость хряща, формирующего ушную раковину, отсутствует. Сам слуховой проход сужен. В 20-25 процентах случаев он может вовсе отсутствовать.
У детей процесс формирования твёрдого нёба во время внутриутробного развития остаётся незавершенным. Там, где у здоровых людей наблюдается срединный шов, у больных заболеванием присутствует продольная щель.
Имеет место быть стопа-качалка. Дефект состоит в неправильном расположении таранной, пяточной и ладьевидных костей. Патологию относят к категории плоского больных деформации стопы у детей. Внешняя стопа новорожденного ребёнка отличается отведением пяточного бугра назад. При этом свод стопы может вовсе отсутствовать.
Больным патологией свойственна ненормальная пропорция длин пальцев. В норме большой палец должен являться самым длинным. У новорождённых с синдромом Эдвардса он уступает по длине второму. По мере роста ребёнка эта особенность становится всё более заметной. Возможно сращение пальцев.
Наблюдается изменение формы нижней челюсти. У больных синдромом Эдвардса подбородок сильно втянут. Это происходит из-за недоразвития нижней челюсти.
Первые признаки синдрома Эдвардса
Первые признаки развития патологии заметно еще в процессе внутриутробного развития. Во время беременности наблюдается маленькая плацента, а также многоводие. Подвижность плода снижена. Присутствует только единственная пупочная артерия. Ребенок появляется на свет с массой тела чуть больше 2 кг. Дополнительно может присутствовать пренатальная гипотрофия. В некоторых случаях выявляются и другие признаки синдрома Эдвардса. Так, может быть выявлена с фиксацией при рождении.
Хорошо заметны фенотипические признаки. Дети обладают низким лбом и маленьким ртом. Имеет место быть деформация ушных раковин и выступающий затылок. Присутствует расщелина нёба и верхней губы. Наблюдается птоз и деформация скелета. Присутствует характерная форма черепа. Иногда развивается аномалия ребер и врожденный вывих бедра. На ушах могут присутствовать козелки. Шея у ребенка короткая. Наблюдается косоглазие и косолапость. Почки имеют маленький размер. Наблюдаются скрещенные пальцы кистей. Стопа имеет форму качалки. На коже присутствуют папилломы и .
Симптомы синдрома Эдвардса
Основными симптомами заболевания выступают внешние и внутренние изменения. Они возникают в результате развития патологии. Такие дети имеют волчью пасть и заячью губу. У них присутствует характерный внешний вид, низкая масса тела при рождении. Дополнительно имеет место быть нервно-психические отклонения. В перечень патологий, касающихся функционирования внутренних органов и моторики, входят:
- Проблема, связанная с работой центральной нервной системы. У многих детей наблюдается задержка нервно-психического развития. Имеет место быть умственная отсталость. Наблюдается недоразвитость мозжечка и мозолистого тела. Присутствует сглаживание или полная атрофия мозговых извилин.
- Проблемы с сердечно-сосудистой системой. Перечень патологии достаточно обширна. Так, может присутствовать открытое овальное отверстие, аналогичный артериальный проток и дефект межжелудочковой перегородки.
- Патологии развития мочеполовой системы. У мальчиков наблюдается крипторхизм и . У девочек присутствует недоразвитость яичников и гипертрофия клитора. Вне зависимости от пола имеет место быть патологические изменения почек. Наблюдается удвоение мочеточников.
- Заболевания органов пищеварения. Наблюдается гастроэзофагеальная рефлюксная болезнь. Глотательный и сосательный рефлексы нарушены. Имеет место быть атрезия пищевода или заднего прохода. Возникает нарушение расположения кишечника.
Дополнительно могут присутствовать косоглазие и сколиоз. Наблюдается атрофия мышц.
Причины и профилактика синдрома Эдвардса
Синдром Эдвардса возникает из-за миопического нерасхождения хромосом. Процесс осуществляется на стадии сперматогенеза или гаметогенеза. Практически в 90% случаев заболевание представлено простой трипаносомой. Реже возникают несбалансированные перестройки хромосом или мозаичная разновидность заболевания. Причины появления синдрома Эдвардса обычно обусловлены появлением в генетическом коде лишней материнской хромосомы.
Генетики пока не смогли определить, почему возникает подобная хромосомная мутация. Считается, что такое явление случайно, и никакие внешние факторы на него влияние не оказывают. Однако всё же ряд причин, способствующих хромосомным аномалиям, сегодня выявлен. В перечень входят:
- инфекции половых органов у родителей;
- наследственность;
- возраст родителей больше 45 лет;
- в процессе беременности мать и плод были подвергнуты радиоактивному излучению;
- женщина долгое время употреблял медикаменты, оказывающие влияние на иммунную, эндокринную и репродуктивную системы (аналогичное правило действует и в отношении мужчин);
- имеет место быть длительное употребление табака, наркотиков и алкоголя.
Как и все генетические патологии, синдром Эдвардса не поддается предотвращению при помощи профилактических методов. Чтобы снизить риск возникновения заболевания, родители могут только предварительно пройти диагностические исследования, позволяющие заранее определить вероятность появления мутации. Дополнительно во время беременности за женщиной и плодом должен осуществляться строгий контроль. Нельзя пренебрегать процедурой антенатального скрининга и рядом других исследований.
Лечение синдрома Эдвардса
Нужно учитывать, что только один из десяти детей, родившихся с подобной патологией, доживают до одного года. Потому первоначальное лечение состоит в избавлении от заболеваний, повышающих риск летального исхода. Если имеет место быть атрезия кишечника или анального прохода, используются меры для дефекации. Ребенка кормят через зонд. Если в организме новорождённого наблюдается активные воспалительные или инфекционные процессы, осуществляется терапевтическое лечение.
Если ребёнка всё же удаётся спасти, в последующем может быть выполнено оперативное вмешательство. В первую очередь выполняется устранение волчьей пасти. Дополнительно проходит борьба с пороками сердца. Пупочная и удаляются при помощи оперативных методов.
Проводится и медикаментозная терапия. Ребенку назначают лекарства, которые позволяют избавить его от запоров, повышенной кислотности и метеоризма. Родители должны понимать, что синдром Эдвардса способствует появлению следующих патологий:
- новообразование в почках;
- гипертензия легких;
- воспаление лёгких;
- высокое артериальное давление;
- синусит и фронтит;
- инфекционные заболевания мочеполовой системы.
Из-за большого количества патологических изменений, происходящих во внутренних и внешних органах больного, дальнейший прогноз развития неблагоприятен. Те дети, которые сумеют дожить до года и достичь относительно взрослого возраста, будут иметь явное умственное торможение в развитии. Они не смогут самостоятельно ухаживать за собой и удовлетворять свои естественные требования. За такими пациентами необходимо обеспечить постоянный уход и контроль на протяжении всей их жизни.
Однако дети с отклонениями понимают, когда с ними ласково общаются, играют и утешают. Дети с таким синдромом могут научиться самостоятельно есть, улыбаться. Возможно постепенное обучение другим полезным бытовым навыкам.
Высокое количество патологий внутренних органов, развившихся неправильно, приводит к высокому проценту детской смертности. Если в первые месяцы беременности женщина попала в группу риска по этому заболеванию, медики рекомендует выполнить аборт по медицинским показаниям. Однако окончательное решение остается за женщиной.
После синдрома Дауна, синдром Эдвардса - самое распространённое хромосомное заболевание. Так как прогнозы для деток, страдающих такой патологией, самые неутешительные (летальный исход либо глубокая олигофрения до конца жизни), родителям обязательно нужно знать, что это за отклонение, когда его можно продиагностировать, чтобы своевременно принять верное решение, оставлять такого малыша в утробе или прервать беременность.
Синдром трисомии 18, или синдром Эдвардса - это хромосомное заболевание, которое характеризуется целым комплексом множественных, очень тяжёлых пороков развития. Причиной является образование дополнительной 18-й хромосомы. Таким образом, кариотип больного с синдромом Эдвардса - три 18-тых хромосомы вместо нормальных двух. В 90% случаев это простая трисомия, реже - мозаичная или транслокация.
Формула, по которой читается кариотип синдрома Эдвардса, обозначается следующим образом: 47,ХХ, 18+ (это девочка) и 47,ХY, 18+ (это мальчик).
По страницам истории. Синдром Эдвардса впервые был описан Джоном Эдвардсом в 1960 году.
Причины

Основная причина заболевания - утроенная (а не сдвоенная, как в норме) 18 хромосома в кариотипе зиготы. Однако, что именно провоцирует такую патологию, генетикам до сих пор неизвестно. Тем более, что появляется лишняя хромосома ещё до оплодотворения. Существует мнение среди врачей, что никакие факты, кроме обычной случайности, здесь не виноваты. С другой стороны, существуют факторы риска, которые позволяют выделить в особую группу предполагаемые причины синдрома Эдвардса, которые нужно иметь в виду парам, собирающимся стать родителями.
- Возраст матери более 45 лет. В этом случае риск синдрома Эдвардса составляет 0,7%.
- Наследственность: если в роду уже рождались детки с подобными отклонениями, очень велик риск родить малыша с патологией в кариотипе.
- Вредные привычки остаются в силе даже здесь. Некоторые учёные доказывают, что мозаичная форма синдрома Эдвардса (она наиболее лёгкая) может быть спровоцирована долгим употреблением наркотиков, огромным количеством никотина или алкоголя в организме одного из родителей.
- Длительный приём сильнодействующих лекарств.
- Инфекции половых путей.
- Радиационное облучение очень часто порождает .
Это лишь предполагаемые генетиками причины возникновения трёх 18-тых хромосом вместо двух. За незыблемый постулат принимать их не стоит. Соответственно с ними, факторы риска синдрома Эдвардса - это возраст матери, уже имеющиеся в роду хромосомные заболевания, вредные привычки.
По сравнению с тем же , эта патология - достаточно редкое явление. Многие родители задаются вопросом, как часто ставят синдром Эдвардса: согласно статистике, 1 больной ребёнок рождается на 7 000 здоровых малышей. Так как прогнозы для таких деток совсем неутешительны, о диагнозе лучше узнать заранее.
Любопытный факт. Пока необъяснимо, но девочек с синдромом Эдвардса рождается в 3 раза больше, чем мальчиков.
Симптоматика

Чтобы принять во время беременности правильное решение, оставить малыша с таким заболеванием или нет, родители должны чётко представлять, что собой являют дети с синдромом Эдвардса, когда родятся. Клиническая картина патологии безрадостна и тяжела для сердца матери и отца. Основные симптомы можно заметить уже с момента рождения малыша.
При рождении:
- низкий вес (около 2 кг) и пренатальная гипотрофия при нормальной и даже превышенной длительности беременности;
- асфиксия.
Деформации лица и черепа:
- аномалии черепа, который имеет долихоцефалическую форму, низкий лоб, выступающий затылок;
- очень маленькие ротовое отверстие и нижняя челюсть;
- расщелины нёба и верхней губы;
- узкие и короткие глазные щели;
- косоглазие;
- деформация ушных раковин, их низкое расположение относительно лица, вытянутость в горизонтальной плоскости;
- отсутствие мочки и козелка;
- суженный наружный слуховой проход, в ряде случаев он вообще отсутствует;
- птоз - обвисание кожи;
- короткая шея с характерной воротниковой кожной складкой.
Деформации скелета:
- короткая грудина, из-за чего уменьшены межреберные промежутки, а грудная клетка короче и шире нормальной;
- в 80 % случаев - аномальное развитие стоп: пятки резко выступают, своды провисают (стопы-качалки), большие пальцы ног утолщены и укорочены;
- скрещённые пальцы рук;
- врождённый вывих бедра;
- косолапость.
Патологии внутренних органов:
- пороки сердца и сосудов;
- гипоплазия мозжечка;
- патологии ЖКТ;
- проблемы с мочеполовой системой;
- нарушения в работе ЦНС;
- ярко выраженная умственная отсталость.
Общее развитие:
- трудности с глотанием, сосанием, дыханием.
Ребёнок с синдромом Эдвардса никогда не сможет жить полноценной жизнью, как все остальные. Всю свою короткую жизнь он будет преодолевать множество препятствий, причём совершенно не будет осознавать своей болезни. Труднее всего приходится родителям, которые вынуждены ежедневно наблюдать за развитием патологий и пороков, с которыми появился на свет их кроха. Когда же проводится диагностика данного заболевания?
Вот это да! Генетик Эдвардс, описавший данное заболевание, выделил свыше 130 симптоматических дефектов, характерных для этого синдрома.
Диагностика

Очень большое значение имеет диагностика синдрома Эдвардса, так как данная хромосомная патология - медицинское показание для прерывания беременности. Для выявления заболевания используются следующие методики.
УЗИ и допплерография
Существуют особые признаки синдрома Эдвардса на УЗИ, которые должен заметить врач:
- множественные аномалии развития плода;
- агенезия пупочной артерии;
- малая величина плаценты;
Но всё это косвенные признаки синдрома Эдвардса при беременности, которые должны быть подтверждены данными других исследований.
Стандартный пренатальный скрининг
Предполагает анализ крови на наличие сывороточных маркеров:
- с 11 по 13 недели: βХГЧ и PAPP;
- с 20 по 24 недели: βХГЧ, свободный эстриола, α-фетопротеин.
Если на основании этих анализов беременная попадает в группу высокого риска, ей предлагается проведение дополнительной диагностики.
Инвазивные методы диагностики

Проводятся в исключительных случаях, когда риск рождения малыша с синдромом Эдвардса очень велик. К инвазивной диагностике относятся:
- биопсия хориона;
- последующее кариотипирование плода.
Если заболевание не было вовремя выявлено, и ребёнок с синдромом Эдвардса всё-таки рождается, он подвергается немедленному всестороннему обследованию.
Диагностика после рождения
Обследование больного ребёнка направлено на выявление пороков развития. Новорождённого с синдромом Эдвардса осматривают разные специалисты:
- неонатолог;
- кардиолог;
- невролог;
- хирург;
- ортопед;
- уролог и др.
Самые важные диагностические исследования, которые выполняются ребёнку с синдромом Эдвардса сразу после его рождения:
- эхокардиография;
- УЗИ почек;
- УЗИ органов брюшной полости.
Какими-то особенностями течение беременности при вынашивании ребёнка с синдромом Эдвардса не отличается. Он так же начинает шевелиться в положенные сроки, бывает даже очень активен, никаких дополнительных токсикозов не наблюдается. Поэтому выявить заболевание в этот период может только современная диагностическая аппаратура. Если же малыш всё-таки родился, вся его недолгая жизнь уйдёт на постоянное лечение сопутствующих пороков.
Мнение врачей. Девочек с синдромом Эдвардса рождается больше не потому, что они чаще всего подвержены такой патологии по 18 хромосоме. Причина - в их выживаемости. Считается, что большая часть беременностей мальчиками с таким синдромом заканчивается самопроизвольным абортом или внутриутробной гибелью плода.
Лечение
У таких деток аномалии развития и всевозможные пороки несовместимы с жизнью, поэтому лечение синдрома Эдвардса сводится к симптоматической помощи. Она направлена на поддержание элементарных физиологических функций, улучшение качества жизни и её продление, насколько это возможно. Хирургическая коррекция врождённых патологий, по мнению большинства врачей, является неоправданной и очень рискованной. В первые дни жизни лечение ограничивается такими мероприятиями, как:
- зондовое питание из-за трудностей глотания и сосания;
- длительная ИВЛ, так как имеются проблемы с дыханием.
После выписки (если это возможно) родители должны обеспечить больному малышу:
- организованный, почти профессиональный уход: хорошо, если рядом с ребёнком будет находиться человек (няня) с медицинским образованием;
- полноценное питание;
- регулярное наблюдение со стороны самых разных детских врачей.
Нужно сразу оговориться, что уход за ребёнком с синдромом Эдвардса требует очень большого терпения и сил.
Прогнозы

Родители, ребёнок которых - носитель синдрома Эдвардса, должны знать, какие прогнозы их ожидают. Как уже было сказано выше, они весьма неутешительны:
- продолжительность жизни таких деток очень невелика;
- 60% умирают до 3 месяцев (чаще всего в эту группу попадают мальчики);
- ещё 10% доживают до года;
- до 10 лет доживает только 1% родившихся;
- самая частая причина смерти - нарушения в работе сердца или остановка дыхания;
- все дети с синдромом Эдвардса до конца дней остаются глубокими олигофренами;
- организм таких деток очень ослаблен, подвержен всевозможным болезням, из-за чего они часто болеют инфекциями мочевыводящих путей, конъюнктивитом, средним , пневмониями;
- словесные навыки общения у таких детей очень ограничены;
- они могут реагировать на слова своих родителей;
- некоторые даже улыбаются, распознают и взаимодействуют с воспитателями;
- в состоянии приобрести такие навыки, как самостоятельное кормление и поднятие головки.
Если в период беременности ребёнку был поставлен диагноз синдром Эдвардса, это заставляет родителей принять ответственное решение (сроки диагностики различных патологий плода можно найти в ). Смогут ли они выхаживать тяжело больного, страдающего множественными патологиями ребёнка? Или есть смысл прервать такую беременность? Здесь только сама пара совместными усилиями может найти единственно правильный для них выход.
В 1971г. на Парижской конференции была утверждена специальная номенклатура записи кариотипа человека.
Нормальный кариотип человека :
46,ХХ - женщина; 46, ХУ - мужчина.
Кариотип при полиплоидии :
69,ХХХ; 69,ХХУ - триплоидии;
92,ХХХХ; 92,ХХХУ - тетраплоидии.
Каритип при моносомии:
45,ХО - единственная моносомия, которая возможна у живых людей (синдром Шерешевского-Тернера).
Кариотип при трисомиях по аутосомам :
47,ХХ,+21 или 47,ХУ,+21 - трисомия по 21 хромосоме (синдром Дауна);
47,ХХ,+13 или 47,ХУ.+13 - трисомия по 13 хромосоме (синдром Патау);
47,ХХ.+18 или 47,ХУ,+18 - трисомия по 18 хромосоме (синдром Эвардса).
Кариотип при трисомиях по половым хромосомам :
47,ХХХ - трисомия Х у женщины;
47,ХУУ - трисомия У у мужчины.
47,ХХУ – синдром Клайнфельтера.
Тетрасомии и пентасомии по половым хромосомам :
48,ХХХХ - тетрасомия Х;
49,ХХХХХ - пентасомии Х;
48,ХХХУ; 49,ХХХХУ - варианты синдрома Клайнфельтера;
48,ХУУУ; 49,ХУУУУ - варианты синдрома полисомии У у мужчины.
Кариотип при хромосомных аберрациях :
46,ХХ,del 5p - - делеция короткого плеча 5 хромосомы (синдром крика кошки) у женщины;
46,ХУ,del 4p - - делеция короткого плеча 4 хромосомы (синдром Вольфа-Хиршхорна) у мужчины;
46,Х,i (Xq) - изохромосома Х по длинному плечу у женщины;
46,ХУ,r (18) - радиальная 18 хромосома у мужчины;
45,ХХ, -Д,-У,+ t (Дq, Уq) - cбалансированная робертсоновская транслокация, образованная соединением длинных плеч одной Д и одной У-хромосомы у женщины.
Кариотип при мозаицизме :
45,Х /46,ХХ или 45,Х /46,ХХ - часть клеток имеет нормальный кариотип (46,ХХ) и часть с моносомией Х (45,Х). Речь идет о мозаичной форме синдрома Шерешевского-Тернера;
47,ХХ,+21/ 46,ХХ - мозаичная форма синдрома Дауна.
Патогенез хромосомных болезней.
При хромосомных болезнях, как правило, наблюдается дисбаланс по большому количеству генов. Измененный генотип проявляется в эмбриональном периоде развития. Самые ранние этапы дробления зиготы контролируются веществами, накопленными в яйцеклетке. Затем происходит включение собственных генов зиготы. Всего в эмбриональном периоде работают около 1000генов, отвечающих за разные этапы онтогенеза. Они разбросаны по всем хромосомам. При геномных и хромосомных мутациях нарушается баланс по большому числу генов, в том числе и по генам, регулирующим эмбриональное развитие. Это неизбежно приводит к нарушению гистогенеза и органогенеза. Формируются пороки развития. Чаще нарушения оказываются несовместимыми с жизнью, что приводит к внутриутробной гибели зародыша. Реже рождается ребенок с пороками развития.
От 35до 50% (сейчас пишут до 70%)зародышей человека погибают на стадии бластоцисты, т.е. до имплантации. У большого процента изних наблюдаются хромосомные перестройки. После имплантации суммарный вклад хромосомных аномалий во внутриутробную гибель у человека составляет 45%.Чем раньше прерывается беременность, тем вероятнее, что это обусловлено хромосомным дисбалансом.
Если аборт происходит в первые 2-4недели, то хромосомный дисбаланс наблюдаются у 60-70%абортусов. В Iтриместре -у 50 %, во 2 триместре -у 30%,в 20-27недель -у 7%и, наконец, 6% мертворождений обусловлены хромосомной патологией.
Если нарушения эмбрионального развития совместимы с жизнью, то рождается ребенок с пороками развития.
У 1% живых новорожденных обнаруживаются те илииные хромосомныеболезни.
Клинически хромосомные болезни проявляются синдромами множественных врожденных пороков развития. Практически все они формируются к моменту рождения. Исключения составляют нарушения в формировании половых признаков при дисбалансе половых хромосом. Некоторые симптомы их проявляются в подростковом периоде. Генетики сравнивают хромосомные болезни с пепелищем после пожара. Пожар - это то, что происходит в эмбриональном периоде. К моменту рождения формируется окончательный фенотип (головешки после пожара). Ничего исправить уже невозможно. Можно лишь провести косметическую коррекцию, прооперировать больного с пороком развития (если синдром совместим с жизнью).
Поскольку при ХБ нарушаются ранние этапы эмбрионального развития, то поражаются одновременно многие органы и системы органов. Это делает клиническую картину многих хромосомных болезней похожей. Чем больше дисбаланс хромосом, тем неспецифичнее картина.
Для любой хромосомной болезни характерен полиморфизм, т.к. индивидуальный генотип особей влияет на экспрессию генов.
КЛИНИКО-ЦИТОГЕНЕТИЧЕСКАЯ ХАРАКТЕРИСТИКА НАИБОЛЕЕ РАСПРОСТРАНЕННЫХ ХРОМОСОМНЫХ БОЛЕЗНЕЙ