Функциональный анализ органических соединений лабораторная. Физико-химические методы анализа органических соединений
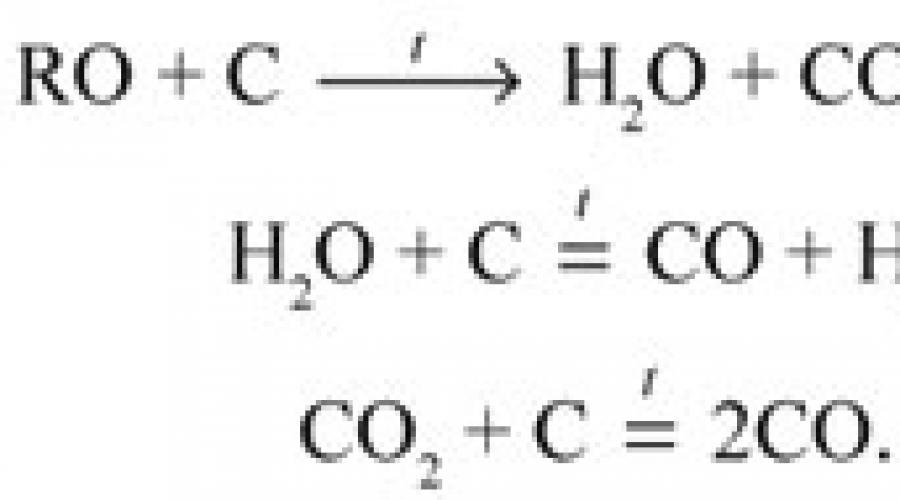
Читайте также
Ю.В.Голубков,
Г.Н.Голубкова
Обнаружение органических веществ
Для поступающих в вузы, учащихся старших классов школ, лицеев, гимназий, студентов колледжей, а также для преподавателей химии
Продолжение. Начало см. в № 10/2010
Глава II.
Элементный анализ органических веществ и определение их строения
Очищенное органическое вещество подвергается качественному и количественному анализу. Наиболее часто в органических соединениях, кроме углерода и водорода, содержатся кислород, азот, сера, галогены, фосфор.
Качественный и количественный анализ органических соединений основан на их разрушении (окислении) с последующим определением обычными методами CO 2 , H 2 O, N 2 , HCl и т.д.
Элементный анализ органических веществ начинался с определения углерода и водорода, поэтому интересно рассмотреть эту тему подробнее.
§ 7. Аналитическое определение углерода и водорода
Сущность метода.
Взвешенное количество исследуемого органического соединения окисляют тем или иным окислителем. Из углерода получают количественно оксид углерода(IV), а из водорода – воду. Массу этих продуктов горения (окисления) определяют весовым путем. Трубка для сожжения (окисления) наполнена таким образом, что другие элементы, находящиеся в органическом соединении, не препятствуют конечному количественному поглощению оксида углерода(IV) и воды в поглотительных аппаратах, содержащих соответствующие поглотители.
1) C + O 2 = CO 2 ,
углекислый газ поглощается баритовой водой (раствор гидроксида бария Ba(OH) 2) и известковой водой (раствор гидроксида кальция Ca(OH) 2), натронной известью (смесь 83 % Ca(OH) 2 , 5 % NaOH и 12 % H 2 O) и другими веществами.
2) H 2 + 1/2O 2 = H 2 O,
вода поглощается безводным перхлоратом магния Mg(ClO 4) 2 (ангидрон); по интенсивности своего осушающего действия он приближается к оксиду фосфора(V), имея перед последним то важное преимущество, что поглотившая воду и расплывшаяся соль магния длительным нагреванием до 220 о С в вакууме может быть вновь обезвожена и повторно использована. Могут использоваться и другие поглотители, например хлорид кальция (от этого соединения пошло название “хлоркальциевая трубка”, см. ниже).
Аппаратура.
Для того чтобы определить принадлежность данного вещества к органическим соединениям, необходимо прежде всего открыть в нем присутствие углерода. Иногда это не представляет затруднений: многие органические вещества при нагревании обугливаются, т.е. превращаются в уголь и этим выдают присутствие в них углерода. Но в целом ряде случаев вещества, содержащие углерод, не обугливаются при накаливании: например, если нагревать спирт, он просто испарится, а если он загорится, то сгорает без копоти. Поэтому наиболее надежным способом открытия углерода в органическом соединении является полное сожжение этого соединения и обнаружение в продуктах горения присутствия оксида углерода(IV).
Для того чтобы сжечь по возможности малое количество вещества и не растерять образующиеся газообразные продукты горения, небольшое количество органического вещества смешивают с окислителем и нагревают. Для этой цели нельзя применять слишком активные окислители, т.к. происходящая при этом реакция затруднит улавливание выбрасываемых из прибора газов. Если, например, смешать сжигаемое вещество с таким сильным окислителем, как хлорат калия KClO 3 (бертолетова соль), то, как известно, при нагревании смеси происходит вспышка, граничащая со взрывом. Поэтому в данном случае применяют малоактивные окислители, действующие лишь при значительном повышении температуры, что позволяет, изменяя температуру, регулировать скорость окисления органического вещества. В органических сожжениях обычно в качестве окислителя применяют оксид меди(II).
Для качественного определения берут несколько миллиграммов исследуемого вещества и смешивают с большим количеством (избыток) зерненного оксида меди(II), смесь насыпают в пробирку, закрывают ее пробкой с газоотводной трубкой и нагревают. Происходит восстановление оксида меди(II) органическим веществом; продукты окисления органического вещества, выходящие по газоотводной трубке, направляют в сосуд с насыщенным раствором гидроксида кальция (известковая вода). Как известно, присутствие оксида углерода(IV) обнаруживается по помутнению известковой воды. Если в состав сожженного вещества входит водород, образующаяся в результате окисления вода осаждается в виде капелек росы на холодных частях прибора и таким образом может быть обнаружена.
Для того чтобы произвести количественное определение углерода и водорода, нужно поглотить чем-нибудь образующиеся продукты горения, каждый отдельно, и взвесить. Принцип работы остается тот же, меняется лишь техника выполнения.
Классический метод сожжения, разработанный в свое время Ю.Либихом, заключается в следующем: сжигаемое вещество не смешивается с оксидом меди(II), а взвешивается в фарфоровой или платиновой лодочке, которая затем помещается в специальную трубку для сожжения (рис. 1). Это длинная стеклянная тугоплавкая трубка, в которую насыпают длинный слой зерненного оксида меди(II), удерживаемый двумя маленькими спиральками, сделанными из свернутой медной сетки. Эти спиральки входят в трубку с легким трением, и поверхность их предварительно окислена. В противоположный конец трубки вставляют более длинную спиральку из окисленной медной сетки так, чтобы между нею и слоем оксида меди(II) оставался промежуток длиннее фарфоровой лодочки с отвешенным веществом.
Приготовленую таким образом трубку помещают в печь для сожжения и при помощи каучуковых пробок со вставленными в них стеклянными трубочками соединяют концы ее с предварительно взвешенными поглотительными приборами: тот конец трубки, в котором находится слой оксида меди(II), соединяется с хлоркальциевой трубкой (рис. 2), в которой происходит поглощение водяного пара, образовавшегося во время горения сжигаемого вещества, а вслед за ней помещают калиаппарат, наполненый концентрированным раствором гидроксида калия и предназначеный для поглощения оксида углерода(IV). На рис. 3 показаны различные конструкции употребляемых калиаппаратов. Противоположный конец трубки для сожжения соединен с газометрами, наполненными: один – воздухом, другой – кислородом (ими пользуются поочередно). Между газометрами и трубкой также помещают поглотительные приборы для удержания влаги и оксида углерода(IV), которые могут содержаться в газах, наполняющих газометры.
Методика определения.
Когда прибор таким образом собран, нагревают до темно-красного каления ту часть трубки, которая наполнена оксидом меди(II), затем вынимают из противоположной части трубки медную спиральку, вводят лодочку с веществом и, снова вдвинув спиральку, закрывают пробкой, устанавливая сообщение с газометром, наполненным воздухом. После этого начинают нагревать длинную спиральку, а затем осторожно повышают температуру той части трубки, в которой находится лодочка с веществом, очень медленно пропуская через трубку струю воздуха.
Вещество, находящееся в лодочке, отчасти испаряется, отчасти разлагается, образуя летучие вещества. Эти горючие пары", смешиваясь с воздухом, увлекаются током его струи к выходу из трубки и, придя в соприкосновение с раскаленным оксидом меди(II), окисляются, восстанавливая оксид меди(II) до металлической меди. Если окисление происходит слишком быстро и образуется слишком много его продуктов (об этом можно судить по количеству пузырьков, проходящих через калиаппарат), понижают температуру и уменьшают интенсивность струи воздуха; в противном случае поступают наоборот.
Если сжигаемое вещество испаряется целиком или разлагается, не образуя угля, в лодочке под конец сожжения ничего не остается. Если же в лодочке образуется уголь, это место трубки нагревают сильнее и вместо воздуха начинают пропускать кислород, в котором уголь постепенно сгорает.
Когда сожжение закончено, через трубку снова пропускают воздух, пока наполняющий трубку и поглотительные приборы кислород не будет вытеснен воздухом. Это необходимо сделать, поскольку поглотительные приборы, когда их взвешивали перед сожжением, были наполнены воздухом, а не кислородом. После этого хлоркальциевую трубку и калиаппарат, дав им принять температуру весов, взвешивают. Привес хлоркальциевой трубки показывает количество поглощенной воды, а привес калиаппарата – количество поглощенного оксида углерода(IV).
Либих производил свои анализы, используя древесный уголь: трубка для сожжения помещалась в корытце и покрывалась слоем горящего угля. Чтобы понизить температуру в каком-нибудь месте трубки, уголь отгребался; чтобы повысить температуру – раздувался ручными мехами. С введением в лабораторную практику светильного газа стали применять печи с большим количеством газовых горелок (рис. 4), пламя которых легко регулировать. В современной практике все чаще и чаще применяют электрические печи.
Либиховский метод органического анализа, называемого иначе элементным анализом (определение элементного состава органического вещества), еще и теперь имеет широкое распространение в лабораториях. Кроме этого метода часто применяют и другие. Например, по методу Денштедта сожжение производится без оксида меди(II); вещество сжигается в струе кислорода в присутствии металлической платины, действующей как катализатор. Это позволяет сократить размеры трубки и уменьшить количество горелок в печи.
В последние годы все большее распространение получает упрощенный метод сожжения Г.Тер-Мейлена и И.Геслинга, заключающийся в том, что анализируемое вещество сжигают в струе кислорода, применяя в качестве катализатора оксид марганца(IV); печь, в которой производится сожжение, очень простой конструкции и нагревается всего двумя горелками. Этот метод удобен еще и тем, что позволяет применять для сожжения очень маленькие количества вещества и является полумикрометодом .
В настоящее время вошли в употребление микрометоды органического анализа. Специальной конструкции микровесы дают возможность взвешивать очень малые количества вещества с точностью до 0,001 мг. Соответствующим образом уменьшены и усовершенствованы трубка для сожжения и поглотительные приборы.
Оригинальный метод органического микроанализа разработан М.О.Коршун и В.А.Климовой. Его сущность заключается в том, что сожжение производится в ничем не наполненной трубке в струе кислорода. Навеску в 3–5 мг вещества помещают в цилиндрический стаканчик из кварца, который вводят в трубку для сожжения (в горизонтальном положении); через трубку пропускают ток кислорода. При нагревании того участка трубки, в котором находится стаканчик со сжигаемым веществом, внутри стаканчика происходит разложение и обугливание вещества, т.к. внутри стаканчика кислород не может циркулировать и потому не находится в избытке. Продукты термического разложения анализируемого вещества, выходя из стаканчика в трубку для сожжения, нагретую до 850–950 °С, встречают большой избыток кислорода и полностью сгорают. Сгорает и оставшийся в стаканчике уголь. Сожжение продолжается всего 10–15 мин. Таким образом, микрометод Коршун и Климовой является скоростным методом.
Если было сожжено а г вещества, привес хлоркальцевой трубки – р г (масса образовавшейся воды), а привес калиаппарата – р 1 г (масса образовавшегося оксида углерода(IV), процентное содержание водорода и углерода в исследуемом веществе определяется следующим образом. 1/9 часть воды по массе приходится на водород, следовательно, в р г воды содержится (р /9) г водорода; чтобы выразить это количество в процентах, следует его умножить на 100 и разделить на навеску: р 100/(9а ) %. В молекуле оксида углерода(IV) 12/44 ее массы приходится на углерод, следовательно, в данном образце содержится (12р 1 /44) г углерода, или 12р 1 100/(44а ) %.
Если сжигаемое вещество состояло из углерода, водорода и кислорода, массу последнего находят по разности (вычитая из 100 % сумму массовых долей водорода и углерода в процентах).
При определении углерода и водорода по методу Унтерцаухера (Унтерцойхера) анализируемое вещество сжигают над оксидом меди(II) в токе воздуха. Водород определяют по количеству выделившейся Н 2 О, которую связывают безводным ВаCl 2 , а углерод – по количеству СО, который образуется при пропускании выделившегося СО 2 над нагретым до 1120 °С углем. При этом содержание СО определяют, как будет описано далее (см. § 10). Метод разработан Й.Унтерцаухером в 1950 г.
В литературе описано еще много вариантов аппаратурного оформления методов определения углерода и водорода в органических соединениях. Обратим внимание на тщательность выполнения методик.
§ 8. Аналитическое определение галогенов
Галоген в органических веществах содержится не в форме иона, поэтому его нельзя осадить нитратом серебра в виде AgCl, AgBr или AgI. Чтобы открыть присутствие галогена в органическом веществе, последнее необходимо разрушить и этим освободить галоген или же перевести его в другое соединение, в котором галоген легко открыть.
Качественное испытание на галоген может быть произведено следующим образом: испытуемое вещество набирают на окисленную медную проволоку или на кусочек оксида меди(II), укрепленный в ушке медной проволоки, и вносят в огонь; органическое вещество сгорает, образуется галогенид меди, окрашивающий пламя в зеленый или голубовато-зеленый цвет. Такой способ открытия галогена известен под названием пробы Бейльштейна . Так можно открыть присутствие галогена, но нельзя решить, какой из них входил в состав исследуемого вещества. Кроме того, реакция не избирательна (мешают нитрилы, некоторые производные пиридина и др.).
Гравиметрические методы.
Для количественного определения галогена в органических соединениях можно поступить следующим образом: в толстостенную стеклянную трубку, запаянную с одного конца, помещают навеску испытуемого вещества, немного нитрата серебра и несколько миллилитров концентрированной азотной кислоты. Трубку запаивают и нагревают до 280–300 °С в специальной печи, поместив предварительно в металлический футляр, на случай, если трубка лопнет, не выдержав давления. В этих условиях органическое вещество сгорает (азотная кислота – окислитель), и в трубке образуется хлорид, бромид или йодид серебра, смотря по тому, какой галоген входит в состав исследуемого вещества. В трубке развивается большое давление вследствие образования оксида углерода(IV) и оксидов азота. По охлаждении трубку вскрывают, извлекают осадок и, промыв и высушив его, взвешивают. Это – метод “мокрого сожжения” Л.Кариуса (1860).
Количественное определение галогена в галогенпроизводных, растворимых в спирте, значительно упростил А.В.Степанов (1906). К спиртовому раствору навески галогенпроизводного добавляют по кусочкам металлический натрий и нагревают в колбе с обратным холодильником. Под влиянием вытесняемого натрием из спирта водорода образуется соответствующий углеводород и галогеноводород, который с натрием дает соответствующую соль, например хлорид натрия. Таким образом галоген переходит в ионное состояние и определяется обычными аналитическими методами (гравиметрически или титриметрически).
Когда производят сожжение веществ, содержащих галоген, с целью определения углерода и водорода, в конце трубки для сожжения помещают спиральку, свернутую из серебряной сетки. Назначение спиральки – удерживать галоген, освобождающийся при сгорании вещества, чтобы он вместе с оксидом углерода(IV) не проник в калиаппарат (поглотительную склянку) и не поглотился щелочью.
По методу “сухого сожжения” вещество сжигают в токе кислорода в кварцевой трубке специального устройства для сожжения, а продукты разложения пропускают над нагретыми платиновыми контактами для того, чтобы обеспечить полное окисление.
По методу сплавления в “бомбе” органическое вещество, содержащее галоген, сплавляют в “бомбе” со смесью нитрата калия, пероксида натрия и тростникового сахара (сахарозы). При этом образуется галогенид натрия, который осаждают нитратом серебра и взвешивают в виде галогенида серебра. Применяемая для сплавления “бомба” (рис. 5) состоит из металлической пробирки глубиной 25 мм и внутренним диаметром 13 мм; она изготовлена из сплава хрома с никелем. Толщина стенок пробирки составляет 1,5 мм; она имеет фланец шириной 3 мм, на который укладывают крышку и свинцовую прокладку. Круглое дно пробирки снабжено небольшой петлей. Крышка “бомбы” прочно закрепляется дугообразным зажимом с плотно пригнанным винтом.
В ы п о л н е н и е а н а л и з а
С м е ш е н и е. Взвешивают в бюксе 300 мг смеси нитрата калия и тростникового сахара (в соотношении 3:1) и 1,5 г пероксида натрия. Одну треть этой смеси загружают в “бомбу” и прибавляют соответствующее количество анализируемого вещества. Затем добавляют остаток смеси, плотно завинчивают крышку “бомбы” и содержимое ее тщательно перемешивают встряхиванием. Такое тщательное перемешивание очень важно для достижения полноты окисления. Многократным постукиванием заставляют содержимое “бомбы” собраться на дне – реакционная смесь готова к сплавлению.
С п л а в л е н и е. “Бомбу” поддерживают щипцами за головку винтового зажима или куском прочной проволоки, вставленной в отверстие головки винта, и постепенно опускают на одну треть в верхнюю часть небольшого бесцветного пламени газовой горелки (следует избегать нагревания “бомбы” слишком близко от ее крышки). Сплавление заканчивается в 10 с; собственно сплавление можно легко распознать по сотрясению “бомбы”. Теоретически сплавление можно считать законченым по истечении указанных 10 с, но рекомендуется держать “бомбу” в пламени дополнительно 5 с, чтобы быть уверенным в полном сплавлении всей реакционной смеси. Затем “бомбу” охлаждают водой под краном.
О х л а ж д е н и е и ф и л ь т р о в а н и е. Прежде всего “бомбу” обмывают снаружи дистиллированной водой. Затем ее вскрывают и горячей дистилированной водой промывают внутреннюю сторону крышки; промывную воду собирают в пробирку для осаждения. Вслед за этим пробирку “бомбы” вставляют в пробирку для осаждения и вновь прибавляют горячую дистилированную воду, чтобы она полностью покрывала пробирку “бомбы”. Для полного растворения реакционной смеси последнюю встряхивают. После растворения всей смеси пробирку поднимают толстой платиновой проволокой и тщательно промывают горячей дистиллированной водой. Затем пробирку берут за петлю щипцами с платиновыми наконечниками и промывают ее внутри горячей дистиллированной водой, которую сливают в первоначальный раствор.
О п р е д е л е н и е х л о р а. Раствор охлаждают в ледяной ванне и подкисляют концентрированной азотной кислотой. Подкисленный раствор фильтруют в другую пробирку для осаждения; к фильтрату прибавляют 1 мл 5%-го раствора нитрата серебра, нагревают на водяной бане и фильтруют осадок.
Осадок хлорида серебра высушивают до постоянного веса и взвешивают, после чего рассчитывают содержание хлора в анализируемом веществе.
О п р е д е л е н и е б р о м а и й о д а. Основной раствор и промывные воды нейтрализируют в присутствии фенолфталеина азотной кислотой до слаборозового окрашивания. Прибавляют 100 мг гидразинсульфата и смесь нагревают на паровой бане 15 мин. После того как эту смесь отфильтруют в другую пробирку для осаждения, ее подкисляют 0,5 мл концентрированной азотной кислоты, прибавляют 1 мл 5%-го раствора нитрата серебра, все вновь нагревают на паровой бане и осадок фильтруют, высушивают и взвешивают (как при определении хлора).
Титриметрические методы.
Определение хлора и брома. Органическое вещество, содержащее хлор и бром, вносят в реакционную колбу и разлагают при 115–125 °С концентрированной серной кислотой в присутствии смеси дихромата калия и дихромата серебра. Сожжение проводят в токе кислорода. Галоген (Х) поступает в приемник, содержащий избыток раствора гидроксида натрия с молярной концентрацией 0,01 моль/л, к которому прибавлен 1 мл пероксида водорода:
2RX -> X 2 + x CO 2 + x H 2 O (окисление),
X 2 + 2NaOH + H 2 O 2 = 2NaX + O 2 + 2H 2 O (поглощение).
Избыток раствора гидроксида натрия оттитровывают раствором кислоты с такой же концентрацией и по количеству израсходованной щелочи вычисляют процентное содержание хлора или брома. Присутствие йода не мешает определению, поскольку он окисляется до йодата. Для летучих веществ или веществ, содержащих также азот или серу, этот метод не пригоден.
Определение йода. Органическое вещество, содержащее йод, сжигают в присутствии катализатора в атмосфере кислорода; при этом образуются йод, оксид углерода(IV) и вода. Йод улавливают и окисляют бромом до образования йодноватой кислоты HIO 3 , последнюю восстанавливают йодоводородом до йода, который определяют йодометрически.
Процес протекает согласно следующим уравнениям:
2RI -> I 2 + x CO 2 + x H 2 O (сожжение),
HIO 3 + 5HI = 3I 2 + 3H 2 O,
I 2 + 2Na 2 S 2 O 3 = 2NaI + Na 2 S 4 O 6 (йодометрическое титрование).
§ 9. Аналитическое определение азота
Качественное определение азота . В продуктах сгорания органических веществ, содержащих азот, последний находится в свободном состоянии. Поэтому, чтобы определить присутствие азота в органическом соединении, его необходимо разрушить и перевести азот в такое соединение, которое легко открыть какими-нибудь качественными реакциями.
Например, по методу Лассеня азот обнаруживают таким образом: в запаянной с одного конца стеклянной трубочке прокаливают немного исследуемого вещества с кусочком металлического калия; при этом вещество обугливается, и из калия, углерода и содержащегося в исследуемом органическом веществе азота образуется цианид калия. Трубочку погружают в небольшое количество воды, вследствие чего она лопается, а цианид калия растворяется в воде. Раствор отфильтровывают от осколков стекла и угля. К полученному раствору, содержащему цианид калия, прибавляют растворы солей железа(II), например сульфат железа(II), и солей оксида железа(III), например хлорида железа(III). Содержащийся в растворе гидроксид калия осаждает гидроксиды железа(II) и (III), а при взаимодействии цианида калия с солями железа(II) образуется гексацианоферрат(II) калия K 4 :
6KCN + FeSO 4 K 4 + K 2 SO 4 .
После этого прибавляют хлороводородную (соляную) кислоту, вследствие чего снова образуются соли железа. После растворения гидроксидов железа(II) и (III) остается осадок образовавшейся из гексацианоферрата(II) калия и хлорида железа(III) берлинской лазури (гексацианоферрата(II) железа(III) Fe 4 3):
3K 4 + 4FeCl 3 = Fe 4 3 + 12KCl.
Образование берлинской лазури (нерастворимой в воде соли характерного синего цвета) свидетельствует о содержании в исследуемом веществе азота.
Количественное определение азота (метод Дюма) . Сжигают органическое вещество, смешанное с оксидом меди(II), пропуская через трубку для сжигания струю углекислого газа; продукты сгорания собирают в прибор, наполненный концентрированным раствором гидроксида калия. Углекислый газ при этом поглощается, объем оставшегося свободного азота измеряют и затем вычисляют его массу.
Когда сжигают органические вещества, содержащие азот, в конце трубки для сжигания помещают восстановленную медную спираль: если образуется незначительное количество оксидов азота, они в соприкосновении с раскаленной медью отдают ей кислород, а азот получается в свободном состоянии.
Выделяющей азот количественно собирают в азотометре (градуированном сосуде) над 50%-м раствором гидроксида калия. Метод разработан Ж.Б.А.Дюма в 1831 г.
Газометрическое определение азота по методу Дюма применимо к органическим соединениям, содержащим азот в любой форме, а именно: к амино-, нитрозо-, азо-, цианосоединениям, алкилнитритам, а также нитратам и гетероциклическим азотсодержащим соединениям.
При определении азота в органических соединениях (главным образом в аминах) по методу Кьельдаля анализируемое вещество разлагают концентрированной H 2 SO 4 в присутствии катализатора, обычно ртути или ее солей. При этом после подщелачивания выделяют аммиак, который отгоняют, поглощают раствором H 2 SO 4 или НCl и определяют титриметрически или колориметрически с реактивом Несслера – щелочным раствором дигидрата тетрайодомеркурата(II) калия K 2 [НgI 4 ] 2H 2 O. Иногда для получения более точных результатов азотсодержащее вещество предварительно обрабатывают восстановителем, например HI. Метод разработан И.Кьельдалем в 1883 г.
Существуют полуавтоматические приборы-анализаторы, позволяющие определять содержание азота одновременно с содержанием углерода и водорода.
§ 10. Аналитическое определение кислорода
Одним из первых методов определения кислорода в органических соединениях является восстановление в токе водорода (гидрирование) при температуре 1120 о С. При этом образуется вода.
Кроме того, органические соединения могут быть разрушены сплавлением с металлическим калием или натрием. При этом кислород образует оксид металла, который легко определить. Однако если в соединении одновременно содержатся кислород и азот, то может образовываться цианат щелочного металла – соль циановой кислоты HNCO.
По методу Унтерцаухера (Унтерцойхера) кислородсодержащее органическое соединение подвергают пиролизу в атмосфере азота. Газообразные продукты пропускают над углем, нагретым до 1110 – 1150 °С. При этом образуется оксид углерода(II), реагирующий затем с твердым оксидом йода(V) (йодноватым ангидридом) I 2 O 5:
5CO + I 2 O 5 = 5CO 2 + I 2 .
Выделившийся йод определяют титриметрически с помощью тиосульфата натрия Na 2 S 2 O 3:
2Na 2 S 2 O 3 + I 2 = 2NaI + Na 2 S 4 O 6 .
Этот метод Й.Унтерцаухер разработал в 1940 г.
Некоторые исследователи предпочитают использовать вертикальную Т-образную кварцевую трубку, заполненую углеродом (рис. 6).
Прямое микроопределение кислорода в органических соединениях.
Вещество подвергают восстановительному разложению над углем в инертной атмосфере. При этом весь кислород переходит в оксид углерода(II):

![]()
Большой вклад в развитие метода микроопределения кислорода и других элементов внесла М.О.Коршун. Она предложила вести гравиметрическое определение кислорода по трем факторам:
1) по убыли массы I 2 O 5 (см. предыдущее уравнение);
2) по привесам меди:
Cu + I = CuI 2 ;
3) по привесу аскарита*:
CO 2 + 2NaOH = Na 2 CO 3 + H 2 O.
§ 11. Аналитическое определение серы
Органическое вещество, содержащее серу, окисляют по методу Кариуса “мокрым сожжением” в концентрированной азотной кислоте в запаянной толстостенной стеклянной трубке в присутствии хлорида бария при температуре 280–300 °С. Присутствующая в веществе сера окисляется до серной кислоты, которая затем реагирует с хлоридом бария с образованием сульфата бария:
H 2 SO 4 + BaCl 2 = BaSO 4 + 2HCl.
После охлаждения трубку вскрывают, извлекают осадок и, промыв и высушив его, взвешивают (гравиметрический анализ).
По методу “сухого сожжения” органическое вещество, содержащее серу, но не содержащие галогенов и азота, окисляют в атмосфере кислорода в присутствии платинового катализатора. При этом сера превращается в газообразные оксиды, поглощаемые водным раствором пероксида водорода:
SO 2 + SO 3 + H 2 O 2 + H 2 O = 2H 2 SO 4 .
Таким образом, конечным продуктом окисления серы является серная кислота, которую титруют раствором гидроксида натрия с молярной концентрацией 0,01 моль/л (титриметрический анализ):
H 2 SO 4 + 2NaOН = Na 2 SO 4 + 2H 2 O.
§ 12. Аналитическое определение фосфора и мышьяка
Для определения фосфора органическое вещество разлагают либо сплавлением с твердыми окисляющими смесями, либо обработкой смесью пероксида водорода и концентрированной серной кислоты. При этом находящийся в органическом веществе фосфор превращается в ортофосфорную кислоту, которую затем определяют весовым путем (гравиметрический анализ) в виде фосфоромолибдата аммония, высушенного предварительно эфиром.
Для определения мышьяка органическое вещество окисляют аналогично соединениям, содержащим фосфор. При этом пользуются либо “мокрым сожжением” (метод Кариуса), либо сплавлением в “бомбе”. Образующуюся мышьяковую кислоту H 3 AsO 4 определяют гравиметрически (весовым путем) в виде магниевой соли пиромышьяковой кислоты:
AsO 3- 4 + Mg 2+ + NH 4 + = MgNH 4 AsO 4 ,
2MgNH 4 AsO 4 Mg 2 As 2 O 7 + H 2 O + 2NH 3 ;
либо йодометрически (титриметрический, или объемный, метод):
H 3 AsO 4 + 2HI = H 3 AsO 3 + I 2 + H 2 O,
I 2 + 2Na 2 S 2 O 3 = 2NaI + Na 2 S 4 O 6 .
§ 13. Определение строения органических веществ
Химия – это область научных знаний и практической деятельности, которая развивается за счет синтеза новых соединений и создания материалов на их основе. Главной задачей химика-исследователя после синтеза соединения является установление строения молекулы.
Определение полной структуры молекулы, включая геометрическую и электронную, является сложной задачей, которая до конца решена быть не может. Поэтому детали строения молекулы более или менее сложного соединения можно изучать бесконечно, добавляя к ним все новые и новые сведения.
Рассмотрим физические основы важнейших методов определения структуры молекул органических соединений. Эти методы называют физическими методами исследования. Химические методы доказательства геометрического строения молекулы (геометрической структуры) также не потеряли своего значения.
Во времена возникновения структурной теории А.М.Бутлерова была осознана важная истина о том, что строение молекулы определяет все ее химические свойства, и по сумме химических свойств можно сделать верное заключение о структуре вещества. Эта истина ни в коей мере не потеряла своего значения в век физических методов исследования. Постоянными в химии остаются анализ и синтез; эти понятия составляют, как известно, основу мыслительного процесса, основу всякого познания. Синтезируя все более и более сложные вещества, химик наперед знает исходные фрагменты, по которым формируется структура нового соединения. Поэтому он предполагает структуру заранее. После синтеза остается только ее доказать. Если же определяется структура природного соединения, о которой нет сведений, то сначала изучаются его химические свойства, которые позволяют сделать выводы о наличии тех или иных структурных элементов. Важнейшим шагом здесь является химический анализ – разложение сложной молекулы на осколки, на отдельные “кирпичики”, и установление характера их комбинации в молекуле.
В последние десятилетия химический анализ был дополнен масс-спектральным анализом – физическим методом, основанным на измерении массы заряженных частиц материи, образующихся при бомбардировке сложных молекул мощным потоком электронов. Этот метод по традиции всегда рассматривается вместе с методами оптической спектроскопии. Вышеупомянутые методы позволяют установить наличие отдельных существенных элементов геометрической (ИК-спектроскопия, спектроскопия комбинационного рассеивания) и электронной (электронная спектроскопия, спектроскопия ядерного магнитного резонанса, спектроскопия электронного парамагнитного резонанса, рентгено-электронная спектроскопия и др.) структуры молекулы и сделать представление о ней более полным.
Спектроскопия ядерного магнитного резонанса (ЯМР-спектроскопия) – один из наиболее современных методов исследования строения органических соединений. В дополнение к электронной и колебательной спектроскопии ЯМР-метод, относящийся к резонансной спектроскопии, позволяет решать наиболее тонкие вопросы структурной химии, в частности, вопросы электронного состояния атомов в молекуле. Метод пригоден для исследования молекул, в состав которых входят атомы с нечетным числом протонов или нейтронов. Ядра таких атомов обладают магнитным моментом и являются парамагнитными.
Теоретические основы указанных методов исследования структуры органических соединений сложны и рассматриваются в соответствующих курсах высшей школы.
* Аскарит – асбест, пропитанный расплавленным NaOH. – Прим. ред.
Основы определения разработаны Ф. Преглем. Навеску вещества в 3-5 мг сжигают при температуре 900 °С в токе кислорода, очищенного от водорода, воды и углекислого газа. Очистка водорода от кислорода осуществляется пропусканием газа над платиновым катализатором при 800 °С. Полная очистка от углекислого газа и воды осуществляется пропусканием через безводный перхлорат магния (ангидрон ) и через асбест, пропитанный расплавом едкого натра (аскарит ).
После трубки сжигания поставлены трубки с поглотителями: ангидроном и аскаритом. Привес первого поглотителя соответствует количеству воды, по которому вычисляют содержание водорода в навеске вещества; привес второго поглотителя дает количество углекислого газа, по которому определяют содержание углерода в анализируемом веществе.
Галогены и серу можно определить, разложив вещество по Кариусу. Галогены в виде галогенида серебра – весовым путем или оттитровывая избыток нитрата серебра. Серу определяют в виде сульфата бария. Газы после сжигания навески пропускают над слоем металлической меди, где происходит восстановление оксидов азота до свободного азота. Азот определяют объемным методом по количеству непоглощенного газа.
Определение молекулярной массы
Для определения молекулярной массы соединения часто пользуются методами криоскопии, основанными на законе Рауля. Для этого определяют температуру замерзания растворителя, а затем раствора. Разница прямо пропорциональна числу молекул вещества, растворенного в данной массе растворителя. Молекулярную массу определяют по формуле:
где р - навеска вещества; Р - навеска растворителя; K - криоскопическая константа;
Аналогично в эбуллиоскопическом методе молекулярную массу определяют через разницу между температурами кипения чистого растворителя и раствора.
Для высокомолекулярных соединений описанные выше методы совершенно неприменимы. В этом случае пользуются тремя методами: вискозиметрическим, осмотическим и седиментационным, которые, в свою очередь, неприменимы к веществам с обычной молекулярной массой.
В настоящее время наиболее часто для определения молекулярной массы неизвестного вещества используют масс-спектрометрию.
Способы выделения индивидуальных веществ
Для выделения соединений применяют следующие физические методы: различные виды перегонки – фракционная при атмосферном давлении, в вакууме, в высоком вакууме, молекулярная перегонка, кристаллизация, экстракция, хроматография. Кроме того, имеется много специальных методов, учитывающих специфику функциональной группы.
· Молекулярная перегонка . Для веществ, которые разлагаются при температуре кипения даже в высоком вакууме, используют «молекулярную перегонку». Принцип ее состоит в том, что при сильном разрежении (10 -5 -10 -8 мм рт. ст.) с нагретой поверхности подлежащего перегонке расплавленного вещества молекулы переходят в газовую фазу при температуре намного ниже температуры кипения данного соединения. Пары вещества затем конденсируются на холодной поверхности. Так удается очистить вещества со сравнительно большой молекулярной массой и хрупкой структурой.
· Перегонка с водяным паром . Как известно, вещество кипит при температуре, когда давление его пара равно атмосферному. Если нагревать две несмешивающиеся жидкости, они закипят при температуре, когда суммарное давление пара обеих жидкостей сравняется с атмосферным. В качестве второй жидкости берут воду. Таким образом, перегонку данной смеси жидкостей можно провести ниже 100°С. Количество обоих веществ в дистилляте определяется соотношением произведения давления пара каждого вещества на его молекулярную массу.
· Кристаллизация . Для очистки твердых веществ применяют перекристаллизацию. Метод основан на том факте, что для большинства соединений при охлаждении их растворов растворимость вещества уменьшается.
· Экстракция . Метод разделения, основанный на разнице коэффициентов распределения вещества между двумя несмешивающимися жидкостями.
Хроматография
Ø Хроматография – метод разделения, основанный на разной скорости перемещения концентрационных зон компонентов исследуемой смеси в потоке подвижной фазы (элюента ) относительно неподвижной фазы.
§ По решаемым задачам выделяют препаративную (количественное разделение веществ) и аналитическую хроматографию (обнаружение веществ и количественная и качественная характеристика смесей).
§ По принципам разделения хроматография подразделяется на адсорбционную , распределительную , ионообменную и ситовую .
v Адсорбционная хроматография . Разделяемые вещества должны отличаться по сродству к твердому адсорбенту, являющемуся неподвижной фазой. В качестве адсорбентов обычно применяют оксид алюминия и силикагель. Значительно реже используют активированный уголь, сульфат бария, силикат магния, полиамиды.
Способность веществ адсорбироваться на полярном адсорбенте в значительной степени определяется их полярностью. По способности адсорбироваться вещества с разными функциональными группами можно расположить в следующей последовательности:
RH < ROCH 3 < R-NO 2 < R-N(CH 3) 2 < R-COOCH 3 < R-NH 2 < R-OH < R-CONH 2 < R-COOH.
По полярности, а значит и по элюирующей способности, растворители-элюенты образуют следующий ряд:
H 2 O > CH 3 OH > C 2 H 5 OH > CH 3 COCH 3 > CH 3 COOC 2 H 5 > C 2 H 5 OC 2 H 5 > CHCl 3 > CCl 4 > циклогексан > н -гексан
Элюирование проводят или одним элюентом (смесью элюентов), или последовательно несколькими элюентами, переходя от менее полярного к более полярному, или смесью двух растворителей (последовательно увеличивая концентрацию более полярного).
· Основные варианты адсорбционной хроматографии .
§ Колоночная адсорбционная хроматография . Адсорбент помещают в колонку. Сверху вначале наносится разделяемое вещество, а затем пропускают элюент, который движется под действием силы тяжести или нагнетается под давлением специальным насосом.
Контроль за разделением веществ ведут или физико-химическими методами (УФ-детекция, рефрактометрия), или аналитическими хроматографическими методами.
§ Тонкослойная хроматография (ТСХ) . Сорбент размещается тонким слоем на стеклянной, алюминиевой или пластмассовой подложке.
Слой сорбента может быть незакрепленным или зафиксированным с помощью специальных химических веществ (крахмал, гипс). Пробу вещества наносят в нижней части пластинки, которую затем помещают в бокс с элюентом. Растворитель за счет капиллярных сил подымается по пластинке (восходящая хроматография), производя разделение. В случае трудноразделимых веществ прибегают к двумерной хроматографии, когда вещество вначале элюируют в одном направлении, а затем элюирование проводят в перпендикулярном направлении.
В современных условиях обычно используют промышленно изготовленные пластинки для препаративной или аналитической ТСХ.
· Выявление и характеристика веществ. Окрашенные соединения наблюдаются при хроматографировании непосредственно. Бесцветные вещества необходимо «выявлять» – превращать в окрашенные соединения.
В зависимости от сорбента, закрепителя и природы разделяемого вещества используют различные методы «выявления», например, углеводы обугливают при высокой температуре, в том числе, и после опрыскивания растворами серной кислоты, аминокислоты дают окрашенные продукты после обработки раствором нингидрина. По интенсивности окраски разделяемых соединений специфическими реагентами судят о содержании их в смеси.
Для характеристики веществ применяют термин «хроматографическая подвижность », которая обозначается как R f , – отношение величины пробега зоны вещества к величине пробега элюента.
v Распределительная хроматография . В основе этого варианта хроматографии лежит распределение веществ между подвижной фазой (газ, жидкость) и неподвижной фазой – жидкость, удерживаемая на твердом инертном носителе. Наибольшее распространение получила распределительная хроматография на бумаге и газо-жидкостная хроматография.
§ Бумажная хроматография . Основой хроматографии на бумаге является распределение смеси разделяемых веществ между водой, адсорбированной на бумаге, и растворителем, насыщенным водой. С помощью этого метода удачно проводилось разделение и идентификация аминокислот и моносахаридов. В настоящее время этот вариант хроматографирования утратил свою актуальность.
§ Газо-жидкостная хроматография . Это распределительная хроматография между стационарной жидкой фазой, нанесенной на носитель, и газом (обычно гелий, азот или водород).
Для характеристики разделяемых веществ используют «время удерживания ». Это время от момента ввода смеси в колонку до выхода из колонки и прохождения вещества через соответствующий детектор, например детектор, регистрирующий изменение теплопроводности. Данный вариант является одним из наиболее широко применяемых хроматографических методов, особенно в аналитических целях.
v Ионообменная хроматография . В основе метода лежит распределение заряженных веществ (ионов) между подвижной и неподвижной фазами в зависимости от их сродства к иононым центрам неподвижной фазы.
По природе ионообменника различают катионную и анионную хроматографию. В качестве элюентов широко используется вода, растворы кислот и щелочей, буферные растворы. Наиболее часто ионообменными материалами служат катиониты и аниониты на основе сшитых полимеров, содержащих ионогенные функциональные группы, а также модифицированная целлюлоза.
v Ситовая хроматография (гель-хроматография) . Отличительной чертой гель-хроматографии является то, что в гелях, образованных трехмерными «сшитыми» макромолекулами, имеются поры определенных размеров, в которые входят меньшие по объему из разделяемых молекул и не входят большие. Поэтому, в отличие от адсорбционной хроматографии, в гель-хроматографии первыми проходят сквозь колонку молекулы большего размера, а самые последними – малые. Разделительная колонка заполняется зернами лиофильного или гидрофильного геля. Примерами таких хроматографических материалов могут служить модифицированные природные гелеобразователи – агар, декстрины, сефадексы (сшитые декстраны) и синтетические сита на основе полиакриламида или «сшитого» полистирола.
Берцелиус Й. (1779 1884) – шведский химик. Научные исследования охватывают все главные проблемы химии первой половины XIX века.
Велер Ф. (1800-1882) – немецкий химик. Работы в неорганической и органической химии. Наряду с Ю. Либихом установил изомерию солей гремучей кислоты.
3 Гмелин Л. (1788-1853) – немецкий химик. Издавал справочники по экспериментальным данным, которые выдержали несколько изданий.
Либих Ю. (1803-1873) – немецкий химик. Создатель теории радикалов, основоположник агрохимии. Изучал органические кислоты.
Бутлеров А. (1828-1886) – русский химик, создатель теории строения органических соединений. Предсказал изомерию многих соединений.
Жерар Ш. (1816-1856) – французский химик. Работал у Ю. Либиха, слушал лекции Ж. Дюма. Создал теорию типов. У Ш. Жерара учились многие русские химики.
Шорлеммер К. (1834 – 1892) – немецкий химик-органик. Работал в области алканов, имеет труды по истории химии.
Лоссень В. (1838 - 1905) – немецкий химик. Основные работы связаны с исследованиями алкалоидов, открыл перегруппировку гидроксамовых кислот.
Кариус Л. (1829-1875) – немеццкий химик. Разработал метод определения серы, галогенов и др. элементов в органических соединениях (1860 г.)
Бейльштейн Ф. (1838-1906) – русский химик-органик. Основные работы в области синтеза ароматических соединений. Инициатор и первый составитель многотомного справочника по органическим соединениям (Handbuch der organische Chemie), известного как «Справочник Бельштейна».
Прегль Ф. (1869-1930) – немецкий химик. Основатель микроанализа органических соединений. Нобелевская премия 1923 г.
Рауль Ф. (1830-1901) – французский химик. Основное направление исследований – изучение растворов.
ОРГАНИЧЕСКИХ ВЕЩЕСТВ АНАЛИЗ
(устар.-орг. анализ), качеств. и количеств. определение состава орг. в-в и установление их строения.
При определении качеств. состава орг. в-в используют разнообразные методы, основанные на хим. р-циях, сопровождающихся образованием продуктов с характерными св-вами (цвет, запах, т-ра плавления и др.), и на измерении физ. и физ.-хим. (хроматографич., спектральных и др.) характеристик идентифицируемых соединений.
При количеств, анализе орг. в-в устанавливают кол-во реагента, вступившего в р-цию с определяемыми орг. соед., или измеряют разл. физ. и физ.-хим. характеристики, связанные с кол-вом определяемого соединения.
О. в. а. включает элементный анализ,
структурно-групповой (включая функц. и стереоспецифич.), молекулярный анализ, фазовый анализ
и структурный анализ.
Исторически первыми были разработаны способы элементного анализа орг. в-в (А. Лавуазье, кон. 18 в.), основанные на их окислении и гравиметрич., титриметрич. или газометрич. определении образовавшихся простых соед. отдельных элементов. Первые методы элементного микрохимического анализа
(микроанализа) разработал Ф. Прегль в нач. 20 в. Со 2-й пол. 20 в. для элементного анализа в-в широко применяют автоматич. анализаторы, основанные на сожжении анализируемой пробы орг. в-ва и газохромато-графич. разделении и определении продуктов сожжения. Анализатор снабжают компьютером и автоматич. системой ввода проб.
Изотопный анализ орг. в-в имеет целью определение в них содержания отдельных изотопов, а также определение соотношения одних и тех же орг. соед., содержащих разные или их сочетания. Для этого чаще всего применяют масс-спектрометрию или многократную газо-жидкостную хроматографию (напр., при разделении обычных и дейте-рир. форм метана или бензола). Наиб. эффективна хромато-масс-спектрометрия.
Большинство методов функционального анализа основано на взаимод.
отдельных функц. групп орг. соед. с подходящими реагентами. Такие р-ции бывают избирательными или ограниченно избирательными, т. е. характерны соотв. только для одной или неск. функц. групп.
Чаще всего используют р-ции, связанные с образованием или исчезновением к-т, оснований, окислителей, восстановителей, воды, газов, реже-осадков и окрашенных в-в. Образовавшиеся к-ты и основания определяют кислотно-основным титрованием
в водной или неводной среде. В неводной среде возможно раздельное потенциометрич. титрование к-ты и основания разной силы при совместном присутствии.
В случае окислит.-восстановит. р-ций, скорость к-рых невелика, обычно используют обратное титрование, т. е. оттитровывают избыток реагента. На образовании или поглощении воды в р-циях орг. соед. основано определение мн. функц. групп с помощью Фишера реактива
(см. также Акваметрия).
Методы, основанные на р-циях, к-рые сопровождаются выделением или поглощением газа, используют редко, т. к. измерение объема или давления обычно требует громоздкой аппаратуры.
На образовании осадков основаны гравиметрич. методы определения небольшого числа функц. групп. Малорастворимые соед., используемые в этих случаях, представляют собой, как правило, металлич. карбоновых и сульфоно-вых к-т, соли орг. оснований, комплексные соед. (в т. ч. хелатные).
Образование окрашенных соед. часто достаточно специфично и позволяет избирательно определять функц. группы фотометрич. методами. Получили распространение (особенно в микроанализе) р-ции, приводящие к образованию флуоресцирующих соед., т. к. чувствительность определения функц. группы в этом случае достаточно велика.
Особой разновидностью функцион. анализа считают методы, основанные на предварит. взаимодействии определяемого в-ва с реагентами и определении образовавшегося продукта. Напр., ароматич. после нитрования можно определять полярографически, а р-ция между аминогруппой и нафталинсульфохлоридом позволяет определять флуориметрически.
Ниже приведены примеры наиб. часто применяемых методов функцион. анализа.
Определение активного водорода в спиртах, аминах, амидах, карбоновых и сульфоновых к-тах, меркаптанах и суль-фонамидах основано на их взаимод. с реактивами Гриньяра (обычно с метилмагнийиодидом; см. Церевитинова метод
)или с LiAlH 4 и измерении объема выделившегося метана или водорода соответственно. Активный в ацетилене и его гомологах определяют по р-ции с солями Ag(I), Hg(I) или Cu(I) с послед, титриметрич. определением выделившихся к-т.
Соединения с ненасыщ. углерод-углеродными связями чаще всего бромируют, иодируют или гидрируют. В первых двух случаях непрореагировавший Вг 2 или I 2 определяют иодометрически, а при гидрировании измеряют объем поглощенного Н 2 . Число двойных связей можно установить по р-ции присоединения солей ртути с послед. титрованием выделившейся к-ты.
При определении гидроксильных групп чаще всего применяют с помощью уксусного, фталевого или пиромеллитового ангидрида, избыток к-рого оттитровывают. Можно использовать хлорангидриды к-т. Гидрокси-группы в фенолах обычно титруют р-рами основании в неводной среде. Фенолы легко бромируются и сочетаются с солями диазония, поэтому оттитровывают р-рами Вг 2 или солей диазония либо приливают к исследуемому р-ру бромид-броматную смесь, избыток к-рой устанавливают иодометрически (см. также Фалина реакция).
Углеводы можно определять окислением периодатом натрия и послед. титрованием избытка окислителя или образующихся к-т. Разработаны многочисл. разновидности этого метода (см., напр., Малапрада реакция).
Для определения орг. пероксисоединений (в т. ч. перокси-кислот) чаще всего используют их взаимод. с KI и послед. титрование выделившегося I 2 р-ром Na 2 S 2 O 3 .
Анализ алкоксисоединений заключается во взаимод. анализируемого в-ва с иодистоводородной к-той с образованием алкилиодидов (см. Цейзеля метод).
Последние определяют разными методами - гравиметрически (в виде AgI) или титриметрически ( , кислотно-основное титрование). Аналогично можно определять и карбоновых к-т. Для идентификации С 1 -С 4 -алкоксигрупп образующиеся алкилиодиды превращают в четвертичные аммониевые соед., к-рые анализируют методами тонкослойной или бумажной хроматографии.
Определение эпоксигрупп основано на их р-ции с хлористым водородом с образованием хлоргидринов; по завершении р-ции избыток НСl оттитровывают р-ром щелочи.
Для определения карбонильных соед. (альдегидов и кето-нов) наиб. часто применяют оксимирование, т. е. их превращение в при взаимод. с гидрохлоридом гидроксил-амина; выделившийся в результате р-ции НСl оттитровывают р-ром щелочи (конечную точку титрования устанавливают с помощью индикатора или потенциометрически). Существует большое число модификаций этого метода. Альдегиды можно определять также по р-ции с бисульфитом Na с послед. кислотно-основным титрованием. Реже используют альдегидов ионами Ag + , р-цию с гидразинами и образование оснований Шиффа.
Хиноны восстанавливают хлоридом Ti(III) или сульфатом V(II); избыток восстановителя определяют титриметрически. Хиноны можно определять также иодометрически.
Для определения карбоновых к-т и их солей наиб. часто применяют кислотно-основное титрование в неводных средах.
Для анализа производных карбоновых к-т разработано большое число методов. Ангидриды после их гидролиза до к-т титруют р-рами щелочей. В случае анализа смеси к-ты и ее ангидрида кислотно-основным титрованием определяют сумму обоих в-в, а затем проводят р-цию ангидрида с морфолином или анилином и оттитровывают выделившиеся к-ты. В последнем случае можно также определять избыток основания титрованием р-ром НСl. Аналогично определяют галогенангидриды или их смеси с к-тами. При этом вместо р-ции с аминами часто используют взаимод. галогенангидрида со спиртом с послед. раздельным титрованием своб. карбоновой к-ты и выделившейся галогеново-дородной к-ты р-ром щелочи.
Определение сложных эфиров карбоновых к-т основано на их гидролизе р-ром щелочи, избыток к-рой оттитровывают р-ром к-ты. Малые кол-ва сложных эфиров обычно определяют спектрофотометрически в виде Fе(Ш)-солей гидрокса-мовых к-т, образующихся при взаимод. сложных эфиров с гидроксиламином.
Для определения азотсодержащих орг. в-в предложено большое число методов. Соед., способные восстанавливаться (нитро-, нитрозо-, ), определяют титано- или ванадатометрически: добавляют избыток р-ра соли Ti(III) или V(II) и непрореагировавший восстановитель от-титровывают р-ром соли Fe(III).
Широкое применение при определении находит титрование р-рами к-т (обычно НСlО 4) в неводной среде. Этот метод часто позволяет раздельно определять орг. и неорг. основания в смесях, а также орг. основания разной силы при совместном присутствии. Амины можно определять, как и гидроксипроизводные, по р-ции их ацилирова-ния. Для определения первичных ароматич. аминов часто используют титрование р-ром в кислой среде, сопровождающееся образованием диазосоединения. Аналогичное титрование вторичных аминов приводит к их N-нитрозирова-нию и также применяется в анализе. При микроанализе первичных ароматич. аминов образовавшиеся диазосоединения обычно подвергают сочетанию с соответствующими азосоставляющими и определяют образовавшийся краситель спектрофотометрически. В случае анализа смесей первичных, вторичных и третичных аминов чаще всего применяют титрование р-ром НСlO 4 в неводной среде исходной смеси (титруются все амины), смеси после ацетилирования уксусным ангидридом (титруются только третичные амины) и смеси после обработки ацетилацетоном или салициловым альдегидом (титруется сумма вторичных и третичных аминов).
Для определения солей арилдиазония р-ром анализируе-мого в-ва титруют навески азосоставляющей (З-метил-1-фенил-5-пиразолона, м-фенилендиамина и др.) или прибавляют к анализируемому р-ру р-р азосоставляющей, избыток к-рой оттитровывают р-ром NaNO 2 в кислой среде. В случае анализа диазосоединений возможно также применение газометрич. анализа, основанного на разложении исследуемого соед. с выделением N 2 , объем к-рого измеряют. Иногда, как и в случае анализа аминов, диазосоединения определяют по р-ции сочетания с послед. спектрофотомет-рич. определением образовавшегося красителя.
Гидразины и обычно оттитровывают иодометри-чески. В случае тиолов можно использовать также взаимод. их с солями серебра или кислотно-основное титрование. Орг. сульфиды окисляют бромид-броматной смесью, избыток к-рой определяют титриметрически.
Широкое распространение для качеств. и количеств. функ-цион. анализа получили также избирательные и достаточно чувствительные методы ИК спектроскопии и ЯМР.
Возникновение стереоспецифического анализа орг. в-в во 2-й пол. 20 в. связано с развитием хроматографич. методов. Для разделения энантиомеров чаще всего предварительно проводят р-цию между анализируемыми в-вами и оптически активными реагентами с образованием диасте-реомеров, к-рые затем разделяют методами газо-жидкост-ной или высокоэффективной жидкостной хроматографии на колонках с оптически активными неподвижными фазами.
Молекулярный анализ орг. в-в основан гл. обр. на применении хроматографии и разл. спектральных методов, к-рые позволяют устанавливать строение орг. соединений.
Фазовый анализ, позволяющий качественно и количественно анализировать кристаллич. формы орг. соед., проводят с помощью рентгенографии
и электронографии. Рентгеновский, структурный анализ
позволяет устанавливать с высокой точностью структурную ф-лу орг. в-ва, определить длины связей между атомами и углы между связями.
Перечисленные выше методы анализа основаны на прямом определении анализируемых в-в или полученных из них производных. В О. в. а. часто применяют также косвенные методы. Так, напр., карбоновые к-ты можно выделить из анализируемой смеси в виде труднорастворимых серебряных или др. солей и затем методом атомно-абсорбц. спектроскопии или рентгено-флуоресцентного анализа определить кол-во соответствующего металла; по результатам такого анализа можно рассчитать содержание карбоновой к-ты. В жидкостной хроматографии эффективно использование косвенного детектирования разделяемых в-в, при к-ром к подвижной фазе прибавляют активный компонент, образующий с продуктами разделения или с хроматографируе-мыми в-вами легко детектируемые соединения.
Приемы анализа и используемая аппаратура зависят от конкретной задачи О. в. а.: определение основного в-ва смеси, орг. или неорг. примеси в орг. в-вах, орг. примеси в неорг. в-ве или анализ сложной многокомпонентной смеси в-в.
Методы О. в. а. широко используют при разработке технологии пром. произ-ва орг. продуктов и в процессе самого произ-ва для разработки методик анализа сырья, вспомогат. в-в, промежут. продуктов на разных стадиях произ-ва, для контроля производств. процесса, готовой продукции, сточных вод и газовых выбросов, для идентификации примесей в промежуточных и конечных продуктах, а также для разработки аналит. методик, обеспечивающих проведение необходимых кинетич. исследований. Во всех случаях необходимо выбирать оптим. варианты методов анализа и их сочетания в соответствии с требованиями к экспрессности, воспроизводимости, точности и т. п.
При разработке аналит. части нормативно-техн. документации на сырье, вспомогат. материалы и готовую продукцию прежде всего устанавливают минимально необходимое и достаточное число аналит. показателей. К таким показателям относят т-ру плавления, р-римость, содержание осн. в-ва в продукте, к-рое определяют прямым методом (обычно титриметрически с применением потенциометрии) или косвенно, вычитая из массы всего продукта массу примесей, определяемых хроматографич. (чаще всего), электрохим. или спектрофотометрич. методами. При использовании функцион. анализа для определения осн. в-ва обычно выбирают методику, предусматривающую определение этого в-ва по функц. группе, образовавшейся на последней стадии его получения. При необходимости, когда анализируемое в-во получают многостадийным синтезом, его определяют по разным функц. группам. Аналит. методы, выбираемые для анализа сырья и готовой продукции, обязательно должны обладать гл. обр. хорошей воспроизводимостью и точностью.
Методы анализа, применяемые в контроле произ-ва, должны быть экспрессными и непрерывными (напр., редокс-метрия, рН-метрия, ). В основе методик контроля процессов произ-ва орг. в-в часто лежит определение исчезающей функц. группы, т. е. группы, подвергающейся превращению на данной стадии произ-ва, что позволяет точно фиксировать конец соответствующей стадии. При этом широко используют тонкослойную, газо-жид-костную, высокоэффективную жидкостную хроматографию, спектрофотометрию, электрохим. методы, проточно-ин-жекц. анализ.
Для анализа промежут. продуктов произ-ва чаще всего применяют титриметрию, а для анализа реакц. смесей-комплекс хроматографич. и спектральных методов, в т. ч. хромато-масс-спектрометрию, сочетание газовой хроматографии с ИК фурье-спектроскопией.
Большое значение приобрел анализ объектов окружающей среды. При разработке соответствующих методик анализа осн. требования к ним заключаются в высокой чувствительности и правильности идентификации определяемых в-в. Этим требованиям удовлетворяют хромато-масс-спектрометрия с использованием двух и более неподвижных фаз.
В клинич. анализе (анализ крови, мочи, тканей и др. объектов на содержание лек. в-в, метаболитов, стероидов, аминокислот и т. п.) важным является не только чувствительность, точность и экспрессность анализа, но и воспроизводимость его результатов. Когда последнее требование имеет решающее значение, применяют хромато-масс-спектрометрию в стандартных условиях, а также высокопроизводительный проточно-инжекц. анализ и разнообразные ферментные методы, обладающие высокой селективностью.
Лит.:
Губен Вейль, Методы органической химии, т. 2, Методы анализа, пер. с нем. 4 изд., М.. 1963; Черонис Н. Д., Ма Т. С., Микро- и полумикро-методы органического функционального анализа, пер. с англ., М., 1973; Сиггиа С.. Ханна Дж. Г., Количественный органический анализ по функциональным группам, пер. с англ., М. ; 1983. Б.
Я. Колоколов.
Химическая энциклопедия. - М.: Советская энциклопедия . Под ред. И. Л. Кнунянца . 1988 .
Смотреть что такое "ОРГАНИЧЕСКИХ ВЕЩЕСТВ АНАЛИЗ" в других словарях:
Анализ воды метод исследования свойств и качеств воды. Применяется для определения количества различных веществ в составе воды, находящейся в контакте с человеком в промышленных и бытовых целях, либо в научных. Содержание 1 Типы воды для… … Википедия
Анализ почвы совокупность операций, выполняемых с целью определения состава, физико механических, физико химических, химических, агрохимических и биологических свойств почвы. Проводят механический (гранулометрический), химический,… … Википедия
АНАЛИЗ ВОДЫ - производится с целью выяснения качества воды и определения возможности использования ее для снабжения рыбоводных прудов. А. в. проводится четыре раза в год: весной (в период весеннего половодья), в середине лета (июль), осенью (в период осеннего… … Прудовое рыбоводство
анализ - АНАЛИЗ (от греч. analysis разложение, расчленение) процедура реального или мысленного расчленения предмета, явления или процесса, а также их взаимоотношений на составные части, элементы, свойства, функции и подсистемы. Процедурой,… … Энциклопедия эпистемологии и философии науки
Идентификация (обнаружение) компонентов анализируемых в в и приблизительная количеств, оценка их содержания в в вах и материалах. В качестве компонентов м. б. атомы и ионы, изотопы элементов и отдельные нуклиды, молекулы, функц. группы и радикалы … Химическая энциклопедия
Определение содержания (массы, концентрации и т. п.) или количеств. соотношений компонентов в анализируемом образце. Определяемыми компонентами м. б. атомы, молекулы, изотопы, функц. группы, фазы и т. п. (см. Элементный анализ, Молекулярный… … Химическая энциклопедия
Учреждение образования «Брестский государственный университет имени А.С. Пушкина»
Кафедра химии
КУРСОВАЯ
РАБОТА
Методы
исследования органических соединений
Выполнила:
студентка 5 курса,
биологического
факультета
специальности
«Биология. Химия»
очной формы
обучения
Петручик
Ирина Александровна
Научный руководитель:
Боричевский
Александр Иванович
Брест, 2012
Методы исследования органических
соединений
ОГЛАВЛЕНИЕ
ВВЕДЕНИЕ………………………………………………………… …………..
3
-
Классификация методов исследования органических веществ………. 4
Простейшие методы исследования органических веществ
2.1.1 Кристаллизация………………………………………… ……… 6
2.1.2 Возгонка………………………………………………………… . 7
2.1.3 Перегонка……………………………………………………… .. 8
2.1.4 Хроматография…………………………………………… …. 9-11
2.2 Анализ органических веществ………………………………….. 12-13
-
Физико-химические методы исследования органических веществ… 14
3.2 Калориметрия……………………………………………… ……… 17
3.3 Рентгенография и электронография…………………………… 18-19
3.4 Электрохимические методы исследования…………………… 20-21
3.5 Спектроскопия…………………………………………… …….. 22-27
ЗАКЛЮЧЕНИЕ…………………………………………………… ……….…. 28
СПИСОК ИСПОЛЬЗОВАННОЙ ЛИТЕРАТУРЫ…………………………. 29
ВВЕДЕНИЕ
Изучение органических веществ
преследует цель установления строения
вещества, его пространственной структуры
и характеристических молекулярных орбиталей,
изучение взаимодействия атомов и молекул,
исследование скоростей и механизмов
реакции. Ввиду огромного числа разнообразных
органических соединений нельзя выработать
единую схему анализа, как часто делается
в неорганическом количественном анализе.
И все же систематическое исследование
позволяет достаточно надежно и быстро
идентифицировать органическое вещество.
Установление строения органического
вещества – это главная цель их изучения
вне зависимости от метода исследования.
Однако интересы, связанные с исследованием
того или иного органического соединения,
уже имеют разный характер. Особенную
важность имеют вопросы, касающиеся природных
ресурсов нашей планеты. Мы знаем, что
особенное значение для человечества
имеют источники нефти и газа, но они ограничены.
Поэтому назрела проблема поисков нового
сырья для органического и нефтехимического
синтеза, получения нефти и газа искусственным
путем. Но это лишь одна из причин изучения
органических веществ. Если посмотреть
вокруг, то все живое на Земле это органическая
химия. Соответственно, изучение органических
веществ это ключ к глобальным открытиям
в области живой природы, возможность
узнать все жизненноважные процессы, найти
пути излечения многих страшных заболеваний,
создавать самим живые материи и т.д.
-
Классификация методов изучения органических веществ.
- простейшие методы изучения: очистка органических веществ (кристаллизация, возгонка, перегонка, хроматография, гель-фильтрация, электрофорез) и анализ органических веществ (количественный и качественные элементные анализы);
- физико-химические методы: рефрактометрия, калориметрия, измерение электрических дипольных моментов, рентгенография и электронография, электрохимические методы (полярография, анодная вольтамперометрия), спектроскопия (фотоэлектронная, масс-спектроскопия, инфракрасная и т.д.)
Простейшие методы исследования органических веществ
-
Очистка органических веществ
Для характеристики чистоты вещества используют следующие константы и методы: температура плавления, температура кристаллизации, температура кипения, коэффициент преломления света, плотность, данные спектров поглощения (коэффициент интенсивности поглощения в электронных и инфракрасных спектрах), данные спектров ядерного магнитного резонанса (ЯМР), масс-спектрометрии, хроматографический анализ, люминесцентный анализ и др.
Получить чистое вещество – означает разделить данную смесь веществ на индивидуальные вещества, очистить до желаемой степени чистоты. Здесь необходимо различать две совокупности методов: методы разделения смеси на компоненты, которые еще не являются чистыми, и методы конечной очистки.
Говоря о чистоте химических веществ, нужно отдавать себе отчет в том, что абсолютно чисто вещество можно представить только теоретически. Абсолютно чистых веществ нет и быть не может. В зависимости от методы очистки вещество содержит определенное количество примесей. Обычными методами очистки можно достичь содержания основного вещества 99,9…99,95%. Специальными методами глубокой очистки можно уменьшить содержание примесей для органических веществ до 10 -3 ….10 -4 %
2.1.1 Кристаллизация
Кристаллизация
является классическим методом очистки
кристаллических веществ. Метод
основан на том, что разные вещества
имеют разную растворимость в определенном растворителе,
причем понижение температуры (за редким
исключением) приводит к уменьшению растворимости
веществ. Фильтрованием горячего раствора
отделяют нерастворимые примести, и после
охлаждения вещество выделяется из раствора
в виде кристаллов. Повторные перекристаллизации
обычно уменьшают количество примесей.
Вариантом метода является кристаллизация
из расплава. Специальный вариант – зонная
плавка – применяется для глубокой очистки
веществ.
Например: нам необходимо очистить салициловую
кислоту от примесей. Для этого мы берем
взвешенную предварительно массу этой
кислоты и рассчитываем необходимый обьем
растворителя – воды, для того, чтобы получить
насыщенный раствор, который впоследствии
можно будет кристаллизировать.
2.1.2 Возгонка (Сублимация)
Многим кристаллическим
веществам свойственна способность
к возгонке, т.е. к переходу в газовую
фазу, минуя жидкую, с последующей
кристаллизацией из газовой фазы.
Этот метод позволяет отделить сублимирующиеся
вещества от несублимирующихся примесей и разделить смесь веществ
с разными температурами сублимации или
температурами кристаллизации из газовой
фазы (градиентная возгонка). Если вещества
возгоняются трудно и при высоких температурах
разлагаются, применяют возгонку в вакууме
или высоком вакууме – до 0,0013 Па (10 -5
мм рт.ст.; 1 мм рт.ст.=133,3 Па). Высоковакуумная
возгонка в различных вариантах применяется
для глубокой очистки.
Очистка твердого вещества
возгонкой возможна только в том
случае, если давление его паров
выше, чем давление паров примесей.
Когда давление паров твердого вещества
соответствует приложенному давлению
получают наилучшие результаты.
Например: Е-стильбен возгоняют при температуре
100 о С и давлении 20 мм рт. ст.
2.1.3 Перегонка
(дистилляция)
Для многих низкоплавких веществ и большинства жидкостей
хорошим методом очистки является
Фракционная перегонка при
условии, что разница в температурах
кипения компонентов смеси достаточно
велика и не образуются азеотропные
смеси. Селективность (эффективность)
фракционной перегонки можно увеличить
специальными приспособлениями: дефлегматорами,
дистилляционными колоннами и др. Для
высококипящих веществ применяется вакуумная
перегонка. Вариантом метода является
перегонка двухкомпонентных систем, которые
при охлаждении расслаиваются, например
перегонка с водным паром: лимонен (т.кип. 178 о С при
760 мм рт. ст.) перегоняется с водой (т.кип.
100 о С при 760 мм рт. ст.) при температуре
98 о С. При этом количественное соотношение
в дистилляте (в граммах) лимонен: вода
составляет 1: 1,54.
2.1.4 Хроматография
Методы хроматографического
разделения основываются на различной
способности веществ адсорбироваться
на поверхности сорбента или распределяться
между двумя несмешивающимися фазами
(жидкость-жидкость, жидкость-газ), из которых одна фаза (жидкая) находится
на поверхности сорбента. Поэтому различают
разные виды хроматографии, а именно: жидкостную
адсорбционную и распределительную хроматографию,
газовую хроматографию.
Жидкостная адсорбционная хроматография основана на различной способности
веществ сорбироваться на поверхности
сорбента и десорбироваться при пропускании
растворителя – элюента. В Качестве сорбентов
применяют оксид алюминия, кремниевую
кислоту и диоксид кремния (силикагели),
гранулированные полисахариды (декстраны)
или другие полимеры, которые в растворителе
набухают, образуя гранулированный гель
(гель-хроматография).
Жидкостная
распределительная хроматография является разновидностью адсорбционной
хроматографии, в которой сорбент (носитель)
покрыт тонкой пленкой какой-то жидкости.
Элюентом обычно является растворитель,
который не смешивается с жидкостью на
сорбенте. При пропускании элюента происходит
распределение веществ между жидкой фазой
и элюентом. Этот вид хроматографии наиболее
пригоден для разделения веществ, хорошо
растворимых в воде или способных образовывать
растворимые в воде соли. К таким веществам
относятся сахар, аминокислоты, многие
органические красители, большая часть
алкалоидов, моно- и поликарбоновые кислоты,
спирты и т. д.
Пример жидкостной
хроматографии смеси стандартов синтетических
фосфолипидов (1) и образца грубого липддного
экстракта из клеточной мембраны эритроцитов
человека(2) на нормально фазной колонке
при детектировании лазерным светорассеивающим
детектором.НЛ – нейтральные липиды; ФЭ
– фосфатидилэтаноламин; ФС – фосфатидилсерин;
ФХ – фосфатидилхолин; СМ – сфингомиелин.
Газовая
хроматография применяется для разделения
смесей газообразных или легкоиспаряемых
жидких и твердых веществ. Принцип метода
подобен жидкостной хроматографии. Разделяемую
смесь разбавляют газом-носителем (H 2,
N 2 , He) и вводят в адсорбционные колонны.
Газ-носитель является одновременно растворителем
и элюентом. В качестве сорбентов используют
тонкие порошки силикатных материалов,
которые могут быть чистыми (газо-адсорбционная
хроматография) или покрытыми пленкой
нелетучей жидкости (газо-жидкостная хроматография).
Используют также капилляры, покрытые
внутри пленкой нелетучей жидкости (капиллярная
хромотография). Газ-носитель постепенно
десорбирует компоненты смеси и уносит
с собой. Присутствие органических веществ
в газе-носителе и их количество обнаруживается
при помощи специальных детекторов и фиксируется
самописцем. В препаративной хроматографии
газ-носитель затем пропускают через специальные
приемники, в которых органические вещества
улавливают вымораживанием.
Этим методом
можно достичь полного разделения
смеси. При использовании адсорбционных
колонн повышенной мощности метод применяется
как препаративный для разделения
небольших количеств веществ (1….10
г).
Пример
газовой хроматографии: скоростной анализ паров взрывчатых
веществ на поликапиллярной колонке при
температуре 170°С.
Поликапиллярная колонка длиной всего
22 см позволяет за 2.5 минуты обнаружить
и идентифицировать следовые количества
паров взрывчатых веществ: 1 - 2,6-динитротолуол,
2 - 2.4-динитротолуол. 3 - 2,4,6-тринитротолуол,
4 - 3,4,5-трининитротолуол, 5 - 2.3,4-тринитротолуол,
6 - гексоген. 7 - тетрил.
-
Анализ органических веществ
Первой задачей является качественное и количественное определение элементного состава. Затем по данным элементного анализа вычисляют простейшую суммарную формулу, определяют молекулярную массу и вычисляют истинную молекулярную брутто-формулу. И наконец, заключительным этапом является определение молекулярной структуры. Для этой цели используют химические методы (постепенное расщепление, получение производных), а в последнее время все чаще применяют физико-химические методы (масс-спектроскопия, рентгеноструктурный анализ, спектроскопия).
Количественный и качественный элементный анализ
В основе методов анализа лежит полное расщепление органического вещества в результате окисления или другим путем и определение химических элементов известными методами. Углерод определяют в виде СО 2, водород – в виде H 2 О, азот – измерением объема N 2 или определением NH 3 или NaCN (в зависимости от вида расщепления), галогены – в виду галогенид-ионов, серу – в виде сульфат- или сульфид-иона, фосфор в виду фосфат-иона и т.д.
Качественно углерод и водород определяют при нагревании с CuO:
C n H 2n +3nCuO>nCO 2 +nH 2 O+3nCu
И выделяющийся оксиду углерода обнаруживают пропусканием газа в раствор Ba(OH) 2 , а воду обнаруживают визуально на стенках пробирки.
Азот, серу и галогены качественно определяют при сплавлении натрием. Образующиеся NaCN, Na 2 S и галогениды натрия обнаруживают в водном растворе обычными аналитическими реакциями.
Для количественного анализа органических соединений существуют специальные пробы. Раньше обычно применялись установки для макроанализа (навеска образца 0,2 … 0,5 г). В наши дни распространены различные приборы для микроанализа (навеска 0,001…0,01 г), для ультрамикроанализа (навеска 10 -5 ...10 -4 г). Для количественного определения углерода и водорода используют приборы, в которых органическое вещество сжигают в токе кислорода: CO 2 улавливают раствором KOH, а H 2 O – специальным абсорбентом и определяют взвешиванием. Для количественного определения азота используют сожжение вещества при нагревании с CuO и объем выделившегося газа измеряют в азометре над раствором KOH. Галогены и серу количественно определяют сожжением образца в атмосфере кислорода, растворением газов в воде и титрованием галогенид-ионов или сульфат-иона.
Разработаны автоматические микроанализаторы с использованием принципа газовой хроматографии, в которых одновременно определяют углерод, водород, азот и серу.
Молекулярную массу соединения обычно определяют масс-спектрометрически.
-
Физико-химические методы исследования органических веществ
-
Спектральные и другие оптические методы;
Электрохимические методы;
Хроматографические методы анализа.
Группа электрохимических методов анализа, основанная на измерении электрической проводимости, потенциалов и других свойств, включает методы кондуктометрии, потенциометрии, вольтамперометрии и т.д.
Но для того, чтобы точно убедиться в более лучшей эффективности этих методов и их действительном большом практическом значении, рассмотрим для сравнения и другие физико-химические методы.
-
Рефрактометрия
, где n – коэффициент преломления света для D-линии натрия (589нм); M – молекулярная масса вещества; ?? – плотность.
Молекулярная рефракция имеет аддитивные свойства, т.е. молекулярная рефракция молекулы может быть получена суммированием рефракций составных частей молекулы. Такими составными частями являются химические связи и совокупность связей и атомов. Эти рефракции вычислены на основе исследований многих органических соединений и могут быть найдены в справочниках. Например:
R CH4 = 4 R C-H ; R CH3NO2 = 3 R C-H +R C-N +R NO2
Явление преломления света связано с поляризуемостью электронной системы молекул. Под влиянием электромагнитного поля света происходит поляризация молекул, в основном их электронных систем. Чем подвижнее электронная система молекулы, тем больше коэффициент преломления света и молекулярная рефракция.
Исследования молекулярной рефракции могут быть использованы для установления структуры соединения. Так, для изучаемого соединения экспериментально определяют молекулярную рефракцию и сравнивают с рефракцией, полученной суммированием рефракций связей по предполагаемой структурной формуле. Если результаты совпадают, то можно считать структуру доказанной, если нет, то надо искать другую структуру. В некоторых случаях наблюдают сильное увеличение молекулярной рефракции по сравнению с ожидаемой (экзальтация рефракции). Это характерно для сопряженных систем.
Значения молекулярной рефракции химических связей, атомов, молекул и ионов могут быть использованы для качественной оценки их поляризуемости. Поляризуемостью молекулы (иона, связи) называют способность ее к поляризации, т.е. к изменению положения ядер и состояния электронного облака под влиянием внешнего электрического поля. В основном происходит электронная поляризация.
3.2 Калориметрия
Калориметрия
является методом исследования тепловых
эффектов химических реакций и процессов
фазовых переходов (например, плавления,
кристаллизации, возгонки, конденсации).
Процесс (реакцию) проводят в специальных
приборах – калориметрах и количественно
оценивают выделенное или поглощенное
тепло.
Калориметрическим
путем определяют молярные теплоты
сгорания веществ. В свою очередь
теплоты сгорания (W) используют для вычисления
теплоты образования вещества E или стандартной
энтальпии образования?H 0 . Теплота
образования вещества может быть вычислена,
исходя из элементов в атомарном состоянии
или из элементов в «стандартном» состоянии
(углерод в виду графита, газообразный
водород и т.д.), при этом полученные числовые
значения, естественно, отличаются. При
рассмотрении табличных данных на это
надо особенно обращать внимание. Обычно
теплоты образования веществ для процесса
вычисляются из атомов элементов, а?H 0
- из элементов в «стандартном» состоянии.
Например, теплота образования углеводородов
из атомов:
- nS -
] – W, где W – теплота сгорания;
- теплота образования CO 2
(393,5 кДж/моль);
- теплота образования воды
(285,8 кДж/моль); S – теплота атомизации (возгонки)
углерода (графита) (-715 кДж/моль);
- теплота атомизации (диссоциации)
молекулы водорода (-436 кДж/моль).
Чем меньше теплота
сгорания, тем больше теплота образования
соединений одинакового состава.
В основном этот
метод служит для сравнения и
характеристики стабильности и реакционной
способности органических соединений.
3.3 Рентгенография
и электронография
Рентгенографический
метод – рентгеноструктурный
анализ – основан на дифракции
рентгеновских лучей в кристалле
вещества. Рентгеновские лучи
(электромагнитное излучение с длиной
волны 0,1-10 нм) при прохождении через кристалл
взаимодействует с электронными оболочками
атомов. В результате этого взаимодействия
происходит дифракция рентгеновских лучей
и на фотопленке получается дифракционная
картина – пятна или окружности. Из дифракционной
картины при помощи сложных расчетов получают
сведения о размещении молекул в элементарной
ячейке кристалла и о расстояниях между
атомами и углах между химическими связями.
Чем меньше число электронов в атоме, тем
слабее рефлексы рентгеновских лучей.
Поэтому определить местонахождение атомов
водорода весьма трудно.
Электронографический
метод подобен рентгенографическому
и основан на взаимодействии потока
электронов с веществом. Поток электронов при прохождении через
вещество напоминает электромагнитное
излучение с очень небольшой длиной волны
и дает дифракционную картину. Эти дифракционные
картины (электронограммы) можно получить
для веществ в газообразном состоянии
или для очень тонких пленок. Дифракция
электронов обусловлена взаимодействием
электронов с атомными ядрами.
Эти методы структурного
анализа дают возможность определить
полную структуру молекулы – межатомные
расстояния, углы между связями, т.е.
точное пространственное расположение всех атомов молекулы в кристаллической
решетке или в газообразном состоянии.
Методом рентгеноструктурного анализа
определена структура таких сложных природных
веществ, как сахароза, пенициллин, стрихнин,
витамин B 12 , некоторые белки (миоглобин)
и нуклеиновые кислоты.
Из рентгенографических
методов исследования было установлено,
что ковалентный радиус атомов при sp 2 - и sp-гибридизации меняется
в зависимости от типа связи, например
в двойной связи С=С (С sp2 - С sp2)
ковалентный радиус атома углерода С sp2
меньше, чем в связи =С-С (С sp2 - С sp3).
В 1-ом случае он составляет 0,067 нм, во 2-ом
– 0,076 нм, а в случае бензола - 0,0695 нм, т.е.
длина связи также зависит уже от самого
соединения и у каждого соединения длины
связей являются уже индивидуальной характеристикой,
что может пригодиться при идентификации
определенного органического соединения.
3.4 Электрохимические
методы исследования
Электрохимические
методы основаны на зависимости силы
тока от приложенного напряжения при
прохождении тока через раствор в электролизерах
специальной конструкции. В результате
появляются кривые зависимости силы тока
– напряжение (потенциал). Эти вольтамперные
кривые характеризуют процессы, проходящие
на электродах. На каотед происходит электрохимическое
восстановление, а на аноде – электрохимическое
окисление. В зависимости от типа изучаемого
процесса (анодного или катодного) применяются
приборы, отличающиеся между собой соотношением
площадей электродов, материалом электродов
и др
Полярография
В основе полярографического метода лежат катодные
процессы (присоединение электрона к веществу
на ртутном капающем электроде). Принципиальная
схема полярографа очень проста. Он состоит
из капающего ртутного микроэлектрода
с непрерывно обновляющейся поверхностью
и электрода сравнения (ртутный или другой
нормальный электрод). Площадь катода
значительно меньше площади анода, поэтому
решающими в этом случае являются процессы
поляризации катода. Органическое вещество
диффундирует к катоду и принимает электрон,
происходит деполяризация катода. Деполяризация
катода начинается при определенном потенциале
Е выд (потенциал восстановления
или выделения, характерный для данного
деполяризатора. В результате начинается
электролиз и сила тока круто возрастает.
При постепенном увеличении напряжения
устанавливается некоторое стационарное
значение силы тока (предельный ток), которое
уже не зависит от повышения напряжения.
Полярографию
можно использовать для характеристики
процесса:
Метод полярографии
широко используется для определения
концентрации веществ в растворах.
Анодная
вольтамперометрия
В основе этого
метода лежат анодные процессы (окисление
органического соединения на платиновом
или графитовом аноде). С точки
зрения экспериментального осуществления
этот метод подобен полярографии.
Анодную вольтамперометрию используют
для изучения процессов окисления:
Метод используют также для количественных определений веществ в растворах.
3.5 Спектроскопия
В основе спектроскопических
методов лежит взаимодействие вещества
с электромагнитным излучением, что вызывает поглощение
излучения или его эмиссию. Взаимодействие
возможно в очень широком интервале электромагнитных
волн, начиная с?-лучей и кончая радиоволнами.
В зависимости
от области электромагнитного спектра
применяют различные эксперимен тальные методы и приборы.
В органической
химии наиболее часто используются
следующие области электромагнитного
излучения:
- ультрафиолетовая
(УФ) и видимая область спектра,
где поглощается энергия, необходимая
для возбуждения электронов в
молекуле (вид электронной спектроскопии);
- инфракрасная
(ИК) область, где поглощается
энергия, необходимая для изменения
колебательных состояний молекулы
(колебательная спектроскопия);
- область радиочастотного
излучения, где энергия затрачивается
для переориентации спинов ядер (спектроскопия
ядерного магнитного резонанса – ЯМР).
Спектральные
методы применяются с целью идентификации
и установления структуры соединений,
анализа смесей, а также позволяют
следить за ходом химических превращений.
Достоинством спектральных методов является малый расход
вещества (1 мг и менее).
Электронная
спектроскопия
Электронный спектр
возникает при поглощении веществом
ультрафиолетового (длины волн 22-400
нм) и видимого (400-800 нм) излучения. Принципиальной
разницы между этими участками спектра нет, они различаются
лишь тем, что волны длиной 400-800 нм воспринимаются
человеческим глазом, и мы видим вещество
окрашенным.
Под действием
УФ-света происходит возбуждение
молекулы, т.е. переход электронов на
более возбужденный уровень и
перераспределение электронной плотности
в молекуле. Труднее всего возбуждаются
электроны, образующие?-связи, легче –
электроны?-связей и неподеленные пары
электронов.
Отправить свою хорошую работу в базу знаний просто. Используйте форму, расположенную ниже
Студенты, аспиранты, молодые ученые, использующие базу знаний в своей учебе и работе, будут вам очень благодарны.
Размещено на http://www.allbest.ru/
Особенности анализа органических соединений:
Реакции с органическими веществами протекают медленно с образованием промежуточных продуктов.
Органические вещества термолабильны, при нагревании обугливаются.
В основе фармацевтического анализа органических лекарственных веществ лежат принципы функционального и элементного анализа.
Функциональный анализ - анализ по функциональным группам, т.е. атомам, группам атомов или реакционным центрам, которые определяют физические, химические или фармакологические свойства препаратов.
Элементный анализ используют для испытания подлинности органических лекарственных веществ, содержащих в молекуле атомы серы, азота, фосфора, галогенов, мышьяка, металлов. Атомы этих элементов находятся в элементоорганических лекарственных соединениях в неионизированном состоянии, необходимым условием испытания их подлинности является предварительная минерализация.
Это могут быть жидкие, твердые и газообразные вещества. Газообразные и жидкие соединения в основном обладают наркотическим действием. Эффект снижается от F - Cl - Br - I. Йодопроизводные в основном обладают антисептическим действием. Связь C-F; C-I; C-Br; C-Cl является ковалентной, поэтому для фармацевтического анализа ионные реакции используют после минерализации вещества.
Подлинность препаратов жидких галогенпроизводных углеводородов устанавливают по физическим константам (температура кипения, плотность, растворимость) и по наличию галогена. Наиболее объективным является способ установления подлинности по идентичности ИК-спектров препарата и стандартных образцов.
Для доказательства наличия галогенов в молекуле используют пробу Бейльштейна и различные методы минерализации.
Таблица 1. Свойства галогенсодержащих соединений
|
Хлорэтил Aethylii cloridum (МНН Ethylchloride) |
Фторотан 1,1,1-трифтор-2хлор-2-бромэтан (МНН Halothane) |
Бромкамфора 3-бром-1,7,7,триметилбицикло-гептанон-2 |
|
|
Жидкость прозрачная, бесцветная, легко летучая, со своеобразным запахом, трудно растворима в воде, со спиртом и эфиром смешивается в любых соотношениях. |
Жидкость без цвета, прозрачная, тяжелая, летучая, с характерным запахом, мало растворима в воде, смешивается со спиртом, эфиром, хлороформом. |
Белый кристаллический порошок или бесцветные кристаллы, запаха и вкуса, очень плохо растворим в воде, легко в спирте и хлороформе. |
|
|
Bilignostum pro injectionibus Билигност Бис-(2,4,6-трийод-3-карбоксианилид) адипиновой кислоты |
Бромизовал 2-бромизовалерианил-мочевина |
||
|
Белый кристаллический порошок, слабо горького вкуса, практически не растворим в воде, спирте, хлороформе. |
Белый кристаллический порошок или бесцветные кристаллы со слабым специфическим запахом, мало растворим в воде, растворим в спирте. |
Проба Бейльштейна
Наличие галогена доказывается путем прокаливания вещества в твердом состоянии на медной проволоке. В присутствии галогенов, образуются галогениды меди, окрашивающие пламя в зеленый или сине-зеленый цвет.
Галогены в органической молекуле связаны ковалентной связью, степень прочности которой зависит от химического строения галогенпроизводного, поэтому для отщепления галогена перевода его в ионизированное состояние необходимы различные условия. Образовавшиеся галогенид-ионы обнаруживают обычными аналитическими реакциями.
Хлорэтил
· Метод минерализации - кипячение со спиртовым раствором щелочи (учитывая низкую температуру кипения, определение ведут с обратным холодильником).
CH 3 CH 2 Cl+KOH c KCl +C 2 H 5 OH
Образовавшийся хлорид-ион обнаруживают раствором серебра нитрата по образованию белого творожистого осадка.
Сl- + AgNO 3 > AgCl + NO 3 -
Фторотан
· Метод минерализации - сплавление с металлическим натрием
F 3 C-CHClBr + 5Na + 4H 2 O> 3NaF + NaCl + 2NaBr + 2CO 2
Образовавшиеся хлорид- и бромид -ионы обнаруживают раствором серебра нитрата по образованию белого творожистого и желтоватого осадков.
Фторид-ион доказывают реакциями:
Реакция с раствором ализаринового красного и раствором нитрата циркония, в присутствии F- красное окрашивание переходит в светло-желтое;
Взаимодействие с растворимыми солями кальция (выпадает белый осадок фторида кальция);
Реакция обесцвечивания роданида железа (красный).
· При добавлении к фторотану конц. H 2 SO 4 , препарат находится в нижнем слое.
Бромизовал
· Метод минерализации - кипячение со щелочью (щелочной гидролиз в водном растворе), появляется запах аммиака:
· Нагревание с конц. серной кислотой - запах изовалериановой кислоты
Бромкамфора
· Метод минерализации методом восстановительная минерализация (с металлическим цинком в щелочной среде)
Бромид-ион определяют реакцией с хлорамином Б.
Билигност
· Метод минерализации - нагревание с концентрированной серной кислотой: отмечается появление фиолетовых паров молекулярного йода.
· ИК-спектроскопия - 0,001% раствор препарата в 0,1 н растворе натрия гидроксида в области от 220 до 300 нм имеет максимум поглощения при л=236 нм.
Йодоформ
· Методы минерализации:
1) пиролиз в сухой пробирке, выделяются фиолетовые пары йода
4CHI 3 + 5O 2 > 6I 2 + 4CO 2 + 2H 2 O
2) нагревание с конц. серной кислотой
2CHI 3 + H 2 SO 4 > 3I 2 + 2CO 2 + 2H 2 O + SO 3
Доброкачественность (чистота галогенсодержащих углеводородов).
Проверку доброкачественности хлорэтила и фторотана проводят, устанавливая кислотность или щелочность, отсутствие или допустимое содержание стабилизаторов (тимола во фторотане - 0,01%), посторонних органических примесей, примесей свободного хлора (брома во фторотане), хлоридов, бромидов, нелетучего остатка.
1) Хлорэтил: 1. Определяют t кипения и плотность,
2. Недопустимую примесь спирта этилового (реакция образования йодоформа)
2) Билигност: 1. Нагревание с кH 2 SO 4 и образование фиолетовых паров I 2
2. ИК-спектроскопия
3) Фторотан: 1. ИК-спектроскопия
2. t кипения; плотность; показатель преломления
3. не должно быть примесей Cl- и Br-
Количественное определение хлорэтила ГФ не предусматривает, но оно может быть выполнено методом аргентометрии или меркуриметрии.
Метод количественного определения - обратное аргентометрическое титрование по Фольгарду после минерализации (реакцию см. в определении подлинности).
1. Реакция перед титрованием:
фармацевтический лекарственный хлорэтил титрование
NaBr + AgNO 3 > AgBrv+ NaNO 3
2. Реакция титрования:
AgNO 3 + NH 4 SCN > AgSCN v + NH 4 NO 3
3. В точке эквивалентности:
3NH 4 SCN + Fe(NH 4)(SO 4) 2 >
Метод количественного определения - аргентометрическое титрование по Кольтгоффа после минерализации (реакции см. в определении подлинности).
1. Реакция перед титрованием:
3NH 4 SCN + Fe(NH 4)(SO 4) 2 > Fe (SCN) 3 + 2 (NH 4) 2 SO 4
точное количество буровато-красный
2. Реакция титрования:
NaBr + AgNO 3 > AgBrv+ NaNO 3
3. В точке эквивалентности:
AgNO 3 + NH 4 SCN > AgSCNv + NH 4 NO 3
обесцвечивание
Билигност
Метод количественного определения - косвенная йодометрия после окислительного расщепления билигноста до йодата при нагревании с раствором перманганата калия в кислой среде, избыток перманганата калия удаляют с помощью нитрата натрия, а для удаления избытка азотистой кислоты к смеси прибавляют раствор мочевины.
Титрант - 0,1 моль/л раствор натрия титсульфата, индикатор - крахмал, в точке эквивалентности наблюдают исчезновение синей окраски крахмала.
Схема реакции:
t; KMnO 4 +H 2 SO 4
RI 6 > 12 IO 3 -
Реакция выделения заместителя:
КIO 3 + 5KI + 3H 2 SO 4 >3I 2 + 3K 2 SO 4 + 3H 2 O
Реакция титрования:
I 2 +2Na 2 S 2 O 3 > 2NaI+Na 2 S 4 O 6
Йодоформ
Метод количественного определения - обратное аргентометрическое титрование по Фольгарду после минерализации.
Минерализация:
CHI 3 + 3AgNO 3 + H 2 O> 3AgI + 3HNO 3 + CO 2
Реакция титрования:
AgNO 3 + NH 4 SCN > AgSCN v + NH 4 NO 3
В точке эквивалентности:
3NH 4 SCN + Fe(NH 4)(SO 4) 2 > Fe (SCN) 3 v + 2 (NH 4) 2 SO 4
Хранение
Хлорэтил в ампулах в прохладном, защищенном от света месте, фторотан и билигност в склянках оранжевого стекла в сухом прохладном, защищенном от света месте. Бромкамфору хранят в склянках оранжевого стекла в сухом прохладном месте.
Хлорэтил используют для местной анестезии, фторотан для наркоза. Бромкамфору применяют в качестве седативного средства (иногда для остановки лактации). Бромизовал является снотворным средством, билигност применяют в качестве рентгеноконтрастного вещества в виде смеси солей в растворе.
Литература
1. Государственная фармакопея СССР / Министерство здравоохранения СССР. - Х изд. - М.: Медицина, 1968. - С. 78, 134, 141, 143, 186, 373,537
2. Государственная фармакопея СССР Вып. 1. Общие методы анализа. Лекарственное растительное сырье / Министерство здравоохранения СССР. - 11-е изд., доп. - М.: Медицина, 1989. - С. 165-180, 194-199
3. Лекционный материал.
4. Фармацевтическая химия. В 2 ч.: учебное пособие / В. Г. Беликов - 4-е изд., перераб. и доп. - М.: МЕДпресс-информ, 2007. - С. 178-179, 329-332
5. Руководство к лабораторным занятиям по фармацевтической химии. Под редакцией А.П. Арзамасцева, стр.152-156.
Размещено на Allbest.ru
Приложение 1
Фармакопейные статьи
Билигност
Бис-(2,4,6-трийод-З-карбоксианилид) адипиновой кислоты
C 20 H 14 I 6 N 2 O 6 M. в. 1139,8
Описание. Белый или почти белый мелкокристаллический порошок слабо горького вкуса.
Растворимость. Практически нерастворим в воде, 95% спирте, эфире и хлороформе, легко растворим в растворах едких щелочей и аммиака.
Подлинность. 0,001% раствор препарата в 0,1 н. растворе едкого натра в области от 220 до 300 нм имеет максимум поглощения при длине волны около 236 нм.
При нагревании 0,1 г препарата с 1 мл концентрированной серной кислоты выделяются фиолетовые пары йода.
Цветность раствора. 2 г препарата растворяют в 4 мл 1 н. раствора едкого натра, фильтруют и промывают фильтр водой до получения 10 мл фильтрата. Окраска полученного раствора не должна быть интенсивнее эталона № 4б или № 4в.
Проба с перекисью водорода. К 1 мл полученного раствора прибавляют 1 мл перекиси водорода; в течение 10--15 минут не должна появляться муть.
Соединения с открытой аминогруппой. 1 г препарата взбалтывают с 10 мл ледяной уксусной кислоты и фильтруют. К 5 мл прозрачного фильтрата прибавляют 3 капли 0,1 мол раствора нитрита натрия. Через 5 минут появившаяся окраска не должна быть интенсивнее эталона №2ж.
Кислотность. 0,2 г препарата встряхивают в течение 1 минуты с кипящей водой (4 раза по 2 мл) и фильтруют до получения прозрачного фильтрата. Объединенные фильтраты титрую! 0,05 н. раствором едкого натра (индикатор--фенолфталеин). На титрование должно расходоваться не более 0,1 мл 0,05 н. раствора едкого натра.
Хлориды. 2 г препарата взбалтывают с 20 мл воды и фильтруют до получения прозрачного фильтрата. 5 мл фильтрата, доведенные водой до 10 мл, должны выдерживать испытание на хлориды (не более 0,004% в препарате).
Фосфор. 1 г препарата помещают в тигель и озоляют до получения белого остатка. К остатку прибавляют 5 мл разведенной азотной кислоты и упаривают досуха, после чего остаток в тигле хорошо перемешивают с 2 мл горячей воды и фильтруют в пробирку через маленький фильтр. Тигель и фильтр промывают 1 мл горячей воды, собирая фильтрат в ту же пробирку, затем прибавляют 3 мл раствора молибдата аммония и оставляют на 15 минут в бане при температуре 38--40° Испытуемый раствор может иметь желтоватую окраску, но должен оставаться прозрачным (не более 0,0001% в препарате).
Иодмонохлорид. 0,2 г препарата взбалтывают с 20 мл воды и фильтруют до получения прозрачного фильтрата. К 10-мл фильтрата добавляют 0,5 г йодида калия, 2 мл соляной кислоты и 1 мл хлороформа. Хлороформный слой должен оставаться бесцветным.
Железо. 0,5 г препарата должны выдерживать испытание на железо (не более 0,02% в препарате). Сравнение проводят с эталоном, приготовленным из 3,5 мл эталонного раствора Б и 6,5 мл воды.
Сульфатная зола из 1 г препарата не должна превышать 0,1%.
Тяжелые металлы. Сульфатная зола из 0,5 г препарата должна выдерживать испытание на тяжелые металлы (не более 0,001% в препарате).
Мышьяк. 0,5 г препарата должны выдерживать испытание на мышьяк (не более 0,0001 % в препарате).
Количественное определение. Около 0,3 г препарата (точная навеска) помещают в мерную колбу емкостью 100 мл, растворяют в 5 мл раствора едкого натра, доливают водой до метки и перемешивают. 10 мл полученного раствора помещают в колбу емкостью 250 мл, прибавляют 5 мл 5% раствора перманганата калия и осторожно по стенкам колбы, при перемешивании, прибавляют 10 мл концентрированной серной кислоты по 0,5--1 мл и оставляют на 10 минут. Затем прибавляют медленно, по 1 капле через 2--3 секунды, при энергичном перемешивании. раствор нитрита натрия до обесцвечивания жидкости и растворения двуокиси марганца. После этого сразу прибавляют 10 мл 10% раствора мочевины и перемешивают до полного исчезновения пузырьков, смывая при этом со стенок колбы нитрит натрия. Затем к раствору прибавляют 100 мл воды, 10 мл свежеприготовленного раствора йодида калия и выделившийся йод титруют 0,1 н. раствором тиосульфата натрия (индикатор -- крахмал).
1 мл 0,1 н. раствора тиосульфата натрия соответствует 0,003166 г C 20 H 14 l 6 N 2 0 6 , которого в препарате должно быть не менее 99.0%.
Хранение. Список Б. В банках оранжевого стекла, в защищенном от света месте.
Рентгеноконтрастное средство.
Йодоформ
Трийодметан
СНI 3 М.в. 393,73
Описание. Мелкие пластинчатые блестящие кристаллы или мелкокристаллический порошок лимонно-желтого цвета, резкого характерного устойчивого запаха. Летуч уже при обыкновенной температуре, перегоняется с водяным паром. Растворы препарата быстро разлагаются от действия света и воздуха с выделением йода.
Растворимость. Практически нерастворим в воде, трудно растворим в спирте, растворим в эфире и хлороформе, мало растворим в глицерине. жирных и эфирных маслах.
Подлинность, 0,1 г препарата нагревают в пробирке на пламени горелки; выделяются фиолетовые пары йода.
Температура плавления 116--120° (с разложением).
Красящие вещества. 5 г препарата энергично взбалтывают в течение 1 минуты с 50 мл воды и фильтруют. Фильтрат должен быть бесцветным.
Кислотность или щелочность. К 10 мл фильтрата прибавляют 2 капли раствора бромтимолового синего. Появившееся желто-зеленое окрашивание должно перейти в синее от прибавления не более 0,1 мл 0,1 н. раствора едкого натра или в желтое от прибавления не более 0,05 мл 0,1 н. раствора соляной кислоты.
Галогены. 5 мл того же фильтрата, разведенные водой до 10 мл, должны выдерживать испытание на хлориды (не более 0,004% в препарате).
Сульфаты. 10 мл того же фильтрата должны выдерживать испытание на сульфаты (не более 0,01% в препарате).
Зола из 0,5 г препарата не должна превышать 0,1%.
Количественное определение. Около 0,2 г препарата (точная навеска) помещают в коническую колбу емкостью 250--300 мл, растворяют в 25 ли 95% спирта, прибавляют 25 мл 0,1 н. раствора нитрата серебра, 10 мл азотной кислоты и нагревают с обратным холодильником на водяной бане в течение 30 минут, защищая реакционную колбу от света. Холодильник промывают водой, в колбу прибавляют 100 мл воды и избыток нитрата серебра оттитровывают 0,1 н. раствором роданида аммония (индикатор -- железоаммониевые квасцы).
Параллельно проводят контрольный опыт.
1 мл 0,1 н. раствора нитрата серебра соответствует 0,01312 г СНI 3 , которого в препарате должно быть не менее 99,0%.
Хранение. В хорошо укупоренной таре, предохраняющей от действия света, в прохладном месте.
Размещено на Allbest.ru
Подобные документы
Понятие рефракции как меры электронной поляризуемости атомов, молекул, ионов. Оценка показателя преломления для идентификации органических соединений, минералов и лекарственных веществ, их химических параметров, количественного и структурного анализа.
курсовая работа , добавлен 05.06.2011
Основные операции при работе в лаборатории органической химии. Важнейшие физические константы. Методы установления строения органических соединений. Основы строения, свойства и идентификация органических соединений. Синтезы органических соединений.
методичка , добавлен 24.06.2015
Изучение теоретических основ методов осаждения органических и неорганических лекарственных веществ. Анализ особенностей взаимодействия лекарственных веществ с индикаторами в методах осаждения. Индикационные способы определения конечной точки титрования.
курсовая работа , добавлен 30.01.2014
Окислительная димеризация метана. Механизм каталитической активации метана. Получение органических соединений окислительным метилированием. Окислительные превращения органических соединений, содержащих метильную группу, в присутствии катализатора.
диссертация , добавлен 11.10.2013
Рассмотрение реакций, основанных на образовании комплексных соединений металлов и без их участия. Понятие о функционально-аналитической и аналитико-активной группах. Использование органических соединений как индикаторов титриметрических методов.
курсовая работа , добавлен 01.04.2010
Химическое строение - последовательность соединения атомов в молекуле, порядок их взаимосвязи и взаимного влияния. Связь атомов, входящих в состав органических соединений; зависимость свойств веществ от вида атомов, их количества и порядка чередования.
презентация , добавлен 12.12.2010
Изомерия как явление существования соединений, одинаковых по составу, но разных по строению и свойствам. Межклассовая изомерия, определяемая природой функциональной группы. Виды пространственной изомерии. Типы номенклатуры органических соединений.
презентация , добавлен 12.03.2017
Основные методы прогнозирования энтальпий образования органических соединений: методы молекулярной механики и аддитивные методы. Метод Бенсона и метод Татевского. Алкилбензолы и их функциональные производные: галогенбензолы, полифенилы, пиридины.
курсовая работа , добавлен 17.01.2009
Галогенирование ароматических соединений: механизм процесса. Расчет показателей при моно- и дихлорировании органических соединений. Расход реагента при максимальном выходе целевого продукта в сложных реакциях. Подбор подходящего механизма реакций.
реферат , добавлен 15.02.2012
Жизнь как непрерывный физико-химический процесс. Общая характеристика природных соединений. Классификация низкомолекулярных природных соединений. Основные критерии классификации органических соединений. Виды и свойства связей, взаимное влияние атомов.